- •
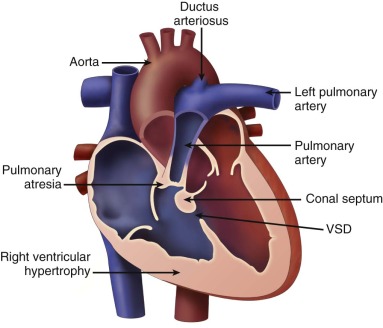
Large malalignment type of ventricular septal defect.
- •
Single semilunar valve, that of the aorta, arising from the heart.
- •
No antegrade flow into the pulmonary artery from the heart
- •
Atretic “platelike” pulmonary valve may be seen or there may be no visible pulmonary valve at all.
- •
Main pulmonary artery segment may be present and small or completely absent.
- •
Branch pulmonary arteries are typically small and may be too small to image and adequately visualize on fetal echocardiography.
- •
Sources of pulmonary blood flow from the aorta, ductus arteriosus, and/or aortopulmonary collaterals should be investigated.
- •
Ductus arteriosus may be absent.
Anatomy and Anatomical Associations
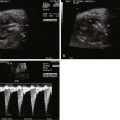
Tetralogy of Fallot with pulmonary atresia (TOF/PA) is a severe form of tetralogy of Fallot (TOF), in which the pathway out of the right ventricle across the pulmonary valve is fully sealed (atretic) ( Figure 11-1 ). Although, in many ways, it is similar to TOF with pulmonary stenosis, this subtype of TOF is unique because the prenatal findings, management, and prognosis can be quite different. Anatomically, there is a large malalignment ventricular septal defect (VSD) in conjunction with an aorta that overrides the ventricular septum. There can be platelike valvar atresia, in which the valve alone is fused, or complete infundibular atresia with the entire subpulmonic region sealed. A main pulmonary artery segment may or may not be present, and oftentimes, just a remnant thread represents the main pulmonary artery. The branch pulmonary arteries and distal pulmonary arterial bed are often abnormal. The aorta is larger than normal.
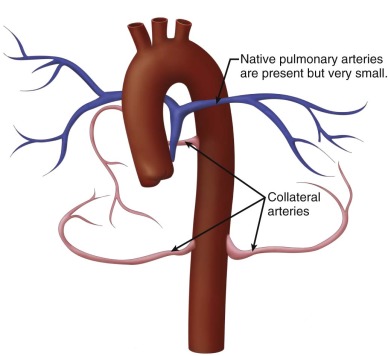
Postnatally, a great deal of variability exists in the origin and distribution of the pulmonary arteries. The branch pulmonary arteries may be supplied by a single ductus arteriosus (DA) and, in most cases, are confluent. Although rare, bilateral ducti can occur with each supplying a single branch pulmonary artery, resulting in discontinuous branch pulmonary arteries. Collateral arterial vessels from the aorta to the pulmonary arteries, referred to as major aortopulmonary collateral arteries (MAPCAs), typically arise from the distal aortic arch or thoracic aorta, but they can also originate from the aortic arch vessels or abdominal aorta ( Figure 11-2 ). Any number of collaterals can be present (typically two to six) with numerous patterns of distribution into the lungs. The size of the collaterals within a single patient can vary as well. Furthermore, the aortopulmonary collateral vessels are residual structures from cardiac development and not intended to serve the same role as a true pulmonary artery. This results in significant abnormalities of these vessels, including diffuse hypoplasia and multiple areas of stenosis. Ductal-dependent pulmonary arteries and MAPCAs can be present in the same patient. As in TOF with pulmonary stenosis, TOF/PA can be associated with other cardiac abnormalities, including coronary artery anomalies, common atrioventricular canal defect, and heterotaxy syndrome.
Frequency, Genetics, and Development
TOF/PA accounts for 20% of all forms of TOF and occurs in 0.07 per 1000 live births. Extracardiac abnormalities are found in one quarter to one half of all infants with TOF/PA with microdeletion of chromosome 22q11 (22q11del) being the most common chromosomal abnormality. 22q11del is present in 90% of patients with DiGeorge syndrome, a genetic disorder associated with multiple extracardiac issues including palatal deformities, feeding and speech difficulties, thymic hypoplasia leading to immunodeficiency and hypocalcemia, varying degrees of developmental, cognitive, and psychological deficits, facial dysmorphism, and renal abnormalities. The incidence of 22q11del in TOF/PA has been reported as high as 40% and is more frequent in the presence of MAPCAs and hypoplastic branch pulmonary arteries. Associated thymic aplasia or a right aortic arch increases the risk of having 22q11del. Other associated genetic conditions include VACTERL (vertebral abnormalities, anal atresia, cardiac abnormalities, tracheoesophageal fistula and/or esophageal atresia, renal agenesis and dysplasia, and limb defects) association, CHARGE (coloboma, heart disease, atresia choanae, retarded growth and retarded development and/or central nervous system anomalies, genital hypoplasia, and ear anomalies and/or deafness) association, Alagille syndrome, and trisomy 21. The precise genetic etiology of TOF/PA remains undefined and, thus far, parallels the mutations described in TOF with pulmonary stenosis.
The development of collateral vessels and an abnormal pulmonary vasculature are what make TOF/PA such a unique anomaly. There is not only the cardiac anomaly of TOF but also a profound change in the makeup of the lung—in essence, a congenital anomaly of the vessels of the lung in association. Early in cardiac development, the lungs normally receive dual blood supply from branches of the dorsal aorta as well as branches from the sixth aortic arches. As normal cardiac development progresses, the branches from the dorsal aorta involute and the branches of the sixth aortic arch enlarge to give rise to main and branch pulmonary arteries. In the absence of antegrade flow across a pulmonary valve as in TOF/PA, normal pulmonary vascular development is interrupted, resulting in the creation of abnormal blood flow patterns to the lungs from the DA or MAPCAs.
Prenatal Physiology
The fetus with TOF/PA is not expected to have significant hemodynamic problems that affect fetal well-being. Atrial level right-to-left shunting is no different than normal. A large VSD allows unrestrictive right-to-left shunting and the combined ventricular cardiac outputs of both right and left ventricle are ejected into the aorta. Pulmonary blood flow is supplied by a DA or collateral vessels originating from the aorta. The fetal lungs receive only approximately 15% to 20% of the combined cardiac output, and therefore, significant hypoplasia or stenoses of the pulmonary vessels may not be evident until the postnatal period when pulmonary blood flow increases significantly. Other physiological features are similar to TOF with pulmonic stenosis. Polyhydramnios and intrauterine growth restriction is common in TOF/PA when associated with 22q11del.
Prenatal Management
Several studies have documented that the degree of right ventricular outflow tract obstruction observed in midgestation can progress, with moderate stenosis advancing to pulmonary atresia by late gestation. Therefore, serial fetal echocardiography is recommended. Identification of whether pulmonary blood flow is supplied by the DA or collateral vessels is important, although origins of all MAPCAs and destination within the lung can be difficult to precisely delineate.
TOF/PA can be categorized into two general subtypes. In the first type, there is platelike pulmonary valvar atresia, a well-formed infundibulum, and a well-formed main pulmonary artery. In this type, the branch pulmonary arteries are typical of what is seen in TOF with pulmonary stenosis, and they are relatively well formed. MAPCAs in this scenario are unusual, and pulmonary blood flow is supplied by the DA. In the second type, there is more profound right ventricular outflow tract atresia, with infundibular and muscular atresia. In this scenario, the main pulmonary can be quite small or absent. MAPCAs are often present and need to be searched for with rigor on fetal echocardiography. A careful search of the ascending and descending aorta to the level beneath the diaphragm using color Doppler imaging should be undertaken. One should look for anomalous vessels arising from the aorta, diving into the lung parenchyma. Pulse Doppler interrogation of these vessels will reveal an arterial waveform.
Because of the high prevalence of associated extracardiac abnormalities, every fetus should have a comprehensive obstetrical ultrasound scan. As in TOF, absent thymus and a right aortic arch increase the risk of 22q11del. Karyotype analysis via chorionic villus sampling or amniocentesis is strongly recommended. Prognosis and counseling will be affected if extracardiac abnormalities are present.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


