Fig. 13.1
Normal neonatal kidney
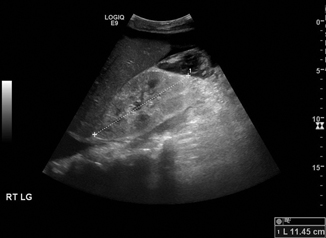
Fig. 13.2
Hyperechoic kidney in congenital nephrotic syndrome. Compare the echogenicity to the adjacent liver
Doppler US can be a useful adjunct in the assessment of vascular problems of the kidney (see below) and is an essential part of the evaluation for renal transplants . Doppler US relies on the frequency shift of blood or other fluid in motion to determine the speed and direction of flow. Flow altering conditions in vessels, such as renal artery stenosis, may be suspected based on alterations in velocity pre- and post-stenosis. Imaging of the native renal vessels may be complicated by overlying bowel gas, and thus fasting beforehand can be helpful [9].
Renal Agenesis and Cystic Dysplasia
Congenital absence of one kidney occurs in approximately 1 in 1000 patients [7]. It is due to agenesis of the Wolffian duct or lack of induction of the metanephros during development. When agenesis is suspected on ultrasound, a careful search for ectopic renal tissue should be made. In females, unilateral renal agenesis may be associated with gynecologic abnormalities such as unicornuate uterus, obstructed hemivagina and ipsilateral renal agenesis (OHVIRA) syndrome, and Mayer–Rokitansky syndrome [10, 11]. Magnetic resonance imaging (MRI) may be indicated if there is a clinical suspicion of other gynecological abnormalities. Forty percent of the patients with a solitary kidney have other urinary tract anomalies such as vesicoureteral reflux (VUR), and a voiding cystourethrogram (VCUG) should be considered [12].
Regardless of whether a solitary kidney is the result of unilateral agenesis, dysplasia, or nephrectomy, long-term follow-up is indicated. The lower limits of acceptable glomerular filtration rate (GFR) in pediatric patients with a solitary kidney, representing two standard deviations below the norm, have been reported to be 78 ml/min/1.73 m2 in children under 2 years, 73 ml/min/1.73 m2 in girls older than 2 and boys 2–13 years old, and 70 ml/min/1.73 m2 in boys older than 13 [13]. Patients falling below these levels should have more in-depth evaluation. Experimental data suggest that decreased nephron mass leads over time to possible hyperfiltration injury and ongoing kidney damage [14]. Patients with a solitary kidney may demonstrate higher blood pressure and increased proteinuria or microalbuminuria [15]. On the other hand, some studies have not found this association [16]. Higher renal vascular resistive indices have also been reported in young children with a solitary kidney, of unclear significance [17]. Thus, although the overall prognosis of patients with a solitary kidney is excellent, they should receive long-term follow-up of blood pressure, proteinuria, and kidney function.
Multicystic dysplastic kidney (MCDK) represents an anomaly of ureteral formation during early development and is the most common neonatal abdominal mass (Fig. 13.3). MCDK is often associated with an atretic or absent ureter [18]. It is characterized by multiple cysts of varying size that do not communicate [19]. There may or may not be visible renal parenchyma. Involution is common and may lead to confusion with renal agenesis if not detected until after involution is complete. Involution appears to occur most rapidly soon after birth. Serial US is an excellent modality for long-term monitoring. The need for nephrectomy in patients whose MCDK does not completely involute is a matter of debate [20]. Increased risk of hypertension has traditionally been used as an argument in favor of nephrectomy, but recent data suggest that this risk is only 5 in 1000, similar to the general population [21].
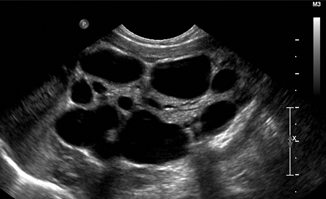
Fig. 13.3
Multicystic dysplastic kidney
Other cystic disorders may be diagnosed by US. Simple renal cysts, which are quite common in adults, are unusual in children. Simple cysts are anechoic without evidence of septae or debris. Cystic changes in the setting of end stage renal disease (ESRD) are common and appear to relate to the duration of dialysis [22]. Annual ultrasound surveillance of pediatric patients on dialysis is prudent due to long-term risk of malignant degeneration. Genetic cystic renal diseases are rare but often suggested by the ultrasound appearance of the kidneys. Identification of multiple cysts in the kidneys should lead to an ultrasonographic survey of other abdominal solid organs, as many cystic renal diseases involve other organs.
Autosomal dominant polycystic kidney disease (ADPKD), the most common genetic cystic disease, generally does not manifest itself until adulthood, but changes may be visible even prenatally. Recently, a fetal pattern of a hyperechoic cortex and hypoechoic medulla has been described with this condition, but the significance of this remains unclear [23]. A few scattered cysts may be evident early in childhood. In a child with a known family history, two or more cysts on ultrasound are diagnostic. Although liver cysts commonly develop, significant hepatic dysfunction is rare. The natural history of this disorder is progression of the cystic disease and development of renal insufficiency in young adulthood [24].
Autosomal recessive polycystic kidney disease (ARPKD) is generally symptomatic early in childhood and may lead to in utero or neonatal death due to pulmonary hypoplasia and Potter sequence. The kidneys are massively enlarged, and the cysts are often small enough that the kidneys appear solid on ultrasound, with loss of corticomedullary differentiation (Fig. 13.4). This condition is associated with hepatic fibrosis and portal hypertension. Perinatal mortality is 30 %, but some patients with a milder phenotype may maintain renal function into adulthood [25]. A variety of rarer pediatric cystic renal diseases exists; a combination of clinical suspicion and genetic analysis may be necessary to diagnose these in the correct clinical setting [26].
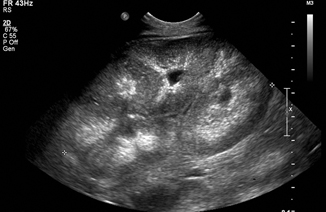
Fig. 13.4
Autosomal recessive polycystic kidney disease (ARPKD) in a neonate. The kidney measures 9 cm (normal for this age is 4.5 cm)
Anomalies of Renal Fusion and Rotation
Anomalies of renal fusion and rotation are relatively common. The kidneys form in the pelvis and rise by differential growth, and failure of ascent leads to ectopic positioning of the kidney. This is commonly in the pelvis below the level of the aortic bifurcation, but may occur at any point along the line of ascent of the kidney. In rare cases, the kidney can be located in the thoracic cavity. This frequently but not exclusively occurs in association with a congenital diaphragmatic hernia [27, 28]. Fusion of the lower pole of the developing kidneys across the midline can lead to horseshoe kidney, which occurs in 1 in 400–500 patients [29]. The ascent of a horseshoe kidney is typically arrested below the inferior mesenteric artery, leading to a low position. It is also possible for both kidneys to ascend ipsilaterally, known as crossed ectopia, and these kidneys may be fused or unfused. In both horseshoe kidney and crossed ectopia, the ureters of each renal moiety drain to their respective sides of the bladder (the ureters originate from the trigone in the normal positions). Malrotation of the pelvis is a common accompanying finding in all forms of renal ectopia, and this may predispose to ureteropelvic junction (UPJ) obstruction [30].
Ultrasound is easily able to identify the location and orientation of ectopic and fused kidneys. The blood supply of these kidneys may be very unusual, with feeding vessels originating at a variety of levels. Doppler US can evaluate the vasculature of the kidneys; however, magnetic resonance angiogram (MRA) or computed tomography angiography (CTA) may be indicated to more precisely delineate the vascular anatomy prior to an operative intervention. Ectopic kidneys have a relatively high incidence of associated urologic abnormalities, including vesicoureteral reflux (VUR) (20–30 % or more), and a VCUG should be considered [31]. Isolated renal ectopia itself is asymptomatic and has no long-term impact on kidney function or hypertension [32].
Duplex Kidney
A duplex kidney is composed of two separate pelvicalyceal systems, with either complete or partial ureteral duplication. This is a common finding, occurring in 1:125 patients. It is a variant of normal anatomy, and patients are often entirely asymptomatic. However, duplex kidneys carry an increased risk for VUR or obstruction. They may be difficult to identify if not associated with dysplasia or hydronephrosis. Suggestive findings on ultrasound include dilated ureter, the presence of a ureterocele in the bladder, or significant hydronephrosis of one pole (Fig. 13.5). Presence of more than one of these features increases the likelihood of a duplex kidney. According to the Weigert–Meyer law, in which the upper pole ureter inserts into the bladder caudally and more medially than the laterally placed lower pole ureter, upper pole moieties are associated with ureterocele and obstruction while lower pole moieties are associated with VUR. A full discussion on the management of duplex kidney is beyond the scope of this chapter.
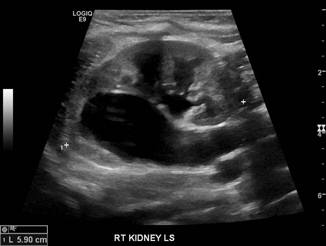
Fig. 13.5
Duplex kidney with upper pole hydroureteronephrosis
Hydronephrosis
Hydronephrosis represents the lion’s share of renal pathology presenting to a pediatric urologist. Antenatal hydronephrosis is very common, and the majority of patients never requires surgical intervention [33]. Evaluation of hydronephrosis is complicated by the fact that the severity can vary according to the patient’s hydration status, and description of hydronephrosis is often subjective and vague. Terms such as “mild,” “moderate,” or “severe” may have very different meanings depending on the observer. The Society of Fetal Urology has established a classification system for the description of hydronephrosis in the neonatal population [34]. This is designed to be used only after the exclusion of VUR, but in practice is commonly used to describe the ultrasound findings without regard to diagnostic status. Grade 0 represents no hydronephrosis, grade 1 only the renal pelvis is seen, grade 2 a few calyces are visualized, and in grade 3 virtually all calyces are dilated. Grade 4 additionally demonstrates cortical thinning. Whenever possible, the Society of Fetal Urology (SFU) criteria or objective measurements such as the APD [35] and should be used to minimize the subjective nature of descriptive terminology.
The pattern of dilation seen on the ultrasound often suggests the underlying etiology. Massive dilation of the renal pelvis with a normal ureter is very suspicious for UPJ obstruction (Fig. 13.6). Follow-up with a diuretic renogram is generally indicated in moderate to severe hydronephrosis in order to confirm the diagnosis, assess the degree of obstruction, and determine differential renal function [36]. Congenital hydronephrosis can often be managed conservatively, but increasing hydronephrosis (particularly with an APD > 3 cm) and deteriorating differential renal function on mercaptoacetyltriglycine (MAG3) renogram indicate UPJ obstruction which requires operative intervention [37].
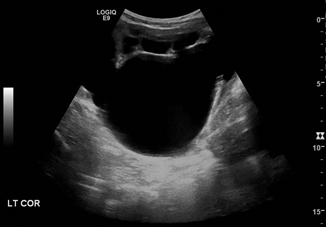
Fig. 13.6
Ureteropelvic junction obstruction with massive hydronephrosis
Hydronephrosis with concomitant hydroureter is suggestive of a congenital megaureter which can be either obstructive or refluxing (Fig. 13.7). Lower grades of VUR that are not associated with dilation are easily missed on US. Formal diagnosis of VUR is typically made with a voiding cystourethrogram (VCUG) or radionuclide voiding cystography (RVC). Both these methods involve exposure to radiation and placement of a urethral catheter. In toilet-trained children, VUR can be diagnosed via indirect radioisotope cystogram using intravenous injection of MAG3.
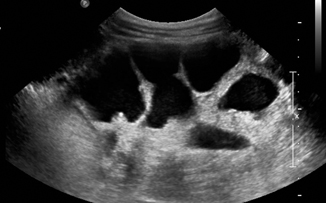
Fig. 13.7
Severe hydronephrosis due to grade V vesicoureteral reflux
Although several groups have attempted to identify US methods of diagnosis that might allow avoidance of both radiation and urethral catheterization, at this date none has been found to be sufficiently sensitive to replace VCUG or RVC [38]. Contrast-enhanced ultrasound has been advocated as a diagnostic tool to avoid radiation exposure. In this technique , a complete renal and bladder ultrasound is performed prior to the instillation of microbubble ultrasound contrast into the bladder via urethral catheter. The US is repeated several times before and after voiding. The presence of microbubbles in the ureter supports the diagnosis of VUR. A grading system has been established corresponding to the traditional VCUG classification of VUR. Microbubbles in the ureter alone represents grade I reflux, while microbubbles in the renal pelvis with hydronephrosis and tortuous ureters represents grade V reflux [39]. VUS is highly operator dependent, does not image the urethra, and the study time is prolonged. Additionally, the cost of ultrasound contrast is much greater than that of fluoroscopic contrast. These factors have limited its widespread adoption, and at this point VCUG remains the gold standard imaging test [40].
Bilateral hydroureteronephrosis in a male infant should raise suspicion of posterior urethral valves (PUV) [41]. The partial urethral obstruction caused by the congenital valves leads to a thick-walled, “keyhole” bladder that can be seen on ultrasound, and careful scanning from the perineal approach may demonstrate the dilated posterior urethra proximal to the valves. Catheter decompression followed by VCUG is the first line of management and should be performed prior to hospital discharge for any infant with suspected valves.
Infection
UTI is very common and represents a significant health care burden; 6.6 % of girls and 1.8 % of boys develop a UTI by age 6 [42]. Despite the frequency of this problem, the indication for imaging and timing of imaging remain a matter of considerable debate in the pediatric urology community, particularly with regard to the use of VCUG after UTI. Although a complete discussion of this topic is beyond the scope of this chapter, a brief discussion of the application of renal sonography to UTI is warranted. UTI can be divided into lower tract infection (cystitis) and upper tract infection (pyelonephritis). In the setting of lower UTI, US may demonstrate thickening of the bladder wall or debris within the bladder. This generally does not add much information to the history and physical exam, urinalysis, and urine culture, and routine use of US to diagnose cystitis cannot be recommended. However, US has the utility in identifying anatomic abnormalities such as hydronephrosis or ureterocele that may require intervention. Recommendations vary as to the indication for and timing of ultrasound after UTI. American Academy of Pediatrics guidelines currently recommend that all infants 2–24 months old with febrile UTI have a screening renal ultrasound performed at some point following treatment [43]. The National Institute for Health and Care Excellence (NICE) guidelines in the UK recommend a renal ultrasound within 6 weeks for infants less than 6 months old with an uncomplicated UTI and imaging during the acute episode for infants with a recurrent UTI or severe illness. For children older than 6 months, US is recommended in the acute setting for seriously ill children and within 6 weeks for a recurrent UTI. No ultrasound is recommended for simple UTI in children older than 6 months [44].
Diagnosis of pyelonephritis as opposed to cystitis may be made based on clinical and radiologic criteria. Pyelonephritis is typically associated with fever and flank pain, with or without symptoms of cystitis [45]. US is often normal in the setting of acute pyelonephritis, but there are several suggestive findings. Inflammation of the renal pelvis and ureter may be seen, and edema of the kidney parenchyma can lead to increased renal volume. Corticomedullary differentiation may be focally lost in the acute setting [46]. If clear documentation of acute pyelonephritis is clinically warranted, a dimercaptosuccinic acid (DMSA) scan to detect renal cortical perfusion abnormalities is indicated [47]. CT may also reveal wedge-shaped hypodensities in contrast uptake due to acute infection. Several investigators have explored the use of Doppler US in the diagnosis of acute pyelonephritis [48, 49]. Doppler US had good ability to detect renal cortical involvement, with 87–89 % sensitivity and 53–92 % specificity but was not as accurate as DMSA overall.
US may reveal layering echoes in the dependent portions of the infected collecting system, which may represent clot, sediment, or pus. These echoes shift with changes in patient position. With clinical evidence of significant infection and echogenic urine on ultrasound, urgent decompression of the pyonephrosis should be considered by nephrostomy tube or internal stenting.
The long-term sequelae of pyelonephritis can include renal scarring, and this risk is increased in the presence of VUR [50]. Although the gold standard investigation for renal scarring is a DMSA scan, several findings on US may suggest the presence of scar. A globally small kidney may represent diffuse renal scarring or alternatively hypoplasia (Fig. 13.8). Focal renal scars may be evident by locally thinned cortical parenchyma, irregular outline, and calyceal dilatation [51]. If clinically indicated, a DMSA scan at 6–12 months post infection can be used to confirm the presence of scarring [47].
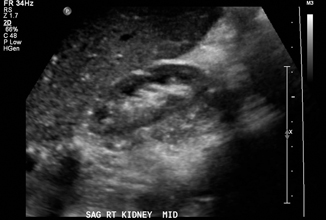
Fig. 13.8
Small globally scarred right kidney in 5-year-old boy with unilateral grade IV vesicoureteral reflux and multiple urinary tract infections
Xanthogranulomatous pyelonephritis (XPN) is a severe chronic renal inflammatory disorder of uncertain origin that is associated with kidney stones, urinary obstruction, and infection [52]. The end result of XPN is a burnt out, nonfunctional kidney characterized by granulomatous inflammatory infiltrate with xanthomatous histiocytes [53]. It is unknown why urinary obstruction due to a stone leads to XPN in some circumstances and not others. Staghorn calculi are common [54]. Although this is usually a process involving the entire kidney, it may occur in a focal form (unassociated with urinary obstruction). Ultrasound may show an enlarged, heterogenous kidney suspicious for a mass. Evidence of urinary obstruction and renal or ureteral stones are often present. Presence of local calcifications may suggest the true diagnosis, but XPN may be mistaken for a neoplastic process. CT scan is generally obtained for better characterization of the process and demonstrates abscess , pyonephrosis, renal atrophy, and inflammatory changes of the perinephric fat or accumulation of perinephric fat. Treatment is generally nephrectomy, although there have been case reports of successful treatment of the focal form of XPN in the pediatric population with long-term intravenous antibiotics [55]. Laparoscopic nephrectomy is being increasingly attempted but the conversion rate to open nephrectomy remains high due to the severe inflammation [56].
Fungal infections of the kidney, particularly candidiasis, are rare but potentially devastating. Preterm infants and immunosuppressed patients are most at risk for fungal involvement of the urinary tract [57]. The classic finding of fungus balls within the collecting system is rare but highly suspicious for candidiasis. These appear as hyperechoic round densities in the collecting system which do not have an acoustic shadow, differentiating them from stones. This finding should prompt an evaluation for systemic fungemia including blood cultures and ophthalmologic exam. In severe cases, bilateral fungal balls or unilateral fungal balls in a solitary kidney may lead to acute renal failure [58]. Fungal obstruction of the bladder outlet has also been described [59]. The mainstay of treatment is systemic antifungal therapy with fluconazole or amphotericin B, but surgical removal of fungal balls may be required in severe or refractory cases [60].
Renal Vascular Disorders
The early onset of severe hypertension in a pediatric patient should raise suspicion of renal artery stenosis (RAS). RAS is usually due to fibromuscular dysplasia in children and as a result commonly involves segmental renal arteries. It is bilateral in 30 % of cases [61]. Other pathologies leading to renal or aortic stenosis include Takayasu’s arteritis and mid-aortic syndrome (stenosis of the abdominal aorta) in neurofibromatosis type I. Angiography remains the gold standard for diagnosis [62], but duplex ultrasound is commonly used as an initial screening test. Doppler interrogation of the renal waveforms suggests stenosis by an elevated peak systolic velocity above 2 m/s or the presence of a tardus et parvus waveform, where the peak systolic velocity is low and slow to reach its maximum. Branch vessel stenoses may be missed if they are distal to the point where the systolic velocities are measured. As branch vessel involvement is common in pediatric RAS, this represents a serious cause of false negative studies. Chhadia and colleagues compared duplex ultrasound in the pediatric population to renal artery angiogram. The sensitivity of US was only 64 %. The majority of patients with missed RAS had branch vessel involvement and severe hypertension, leading the authors to recommend that in the clinical setting of severe hypertension, consideration should be made to proceeding with a diagnostic angiogram despite a negative US study [63]. Endovascular techniques are being increasingly applied to RAS. Angioplasty has been widely used. Stenting has traditionally been avoided in fibromuscular dysplasia as well as in the pediatric population in general, but has been used with some reported success [64]. In the event of unsuitable anatomy or stenosis refractory to endovascular therapy, open repair may be required. Ex vivo repair may be necessary in the case of complex branch vessel stenosis [62].
Renal and Adrenal Neoplasms
US often represents the first-line imaging modality in the diagnosis of abdominal masses . Cystic masses of the kidney are commonly MCDK (discussed above), although cystic nephroma is a rare tumor of the kidney. More commonly, a solid renal tumor may contain cystic components. Antenatal or neonatal detection of a solid renal mass by physical exam or ultrasound should raise the suspicion of a congenital mesoblastic nephroma (CMN). These masses are generally large and often associated with polyhydramnios. The tumor exhibits low echogenicity and often has concentric hyper- and hypoechoic rings. CMN is characterized by infiltration of surrounding renal parenchyma without infiltration of the renal vein. Further imaging with CT or MR is indicated prior to definitive surgical excision, but clear differentiation from a Wilms’ tumor may not be possible preoperatively. These tumors have low malignant potential, except in the case of the cellular form of CMN. Cellular CMN has cytogenetic similarity to congenital fibrosarcoma and may exhibit more aggressive behavior [65].
Wilms’ tumor (WT) appears as a large heterogenous lesion arising from the kidney, without calcifications (although calcifications may be present in 5–10 % of WT). Unlike in CMN, there tends to be a well-defined pseudocapsule [66]. Anechoic components represent necrosis, hemorrhage, or cysts within the tumor. Doppler US is very helpful in the assessment of venous extension of tumor into the renal vein and inferior vena cava. Vessel patency must be interrogated to the level of the hepatic veins. Nephrogenic rests may sometimes be observed; however, on ultrasound these have similar echogenicity to the renal parenchyma and may be missed. Once the diagnosis of tumor is made by US, current Children’s Oncology Group (COG) and Society of Paediatric Oncology (SIOP) protocols mandate appropriate cross sectional imaging (MRI or CT) for further characterization of the tumor and operative planning.
Other rarer renal tumors include clear cell sarcoma of the kidney, rhabdoid tumor, and juvenile renal cell carcinoma. Renal medullary carcinoma is a highly lethal tumor associated with sickle cell trait [65]. US cannot reliably distinguish between these renal tumors. Further imaging and biopsy are generally necessary. Angiomyolipoma is a hamartomatous tumor associated with tuberous sclerosis. The classic defining feature is the presence of fat, which lends a very hyperechoic appearance to the tumor on US. Approximately 33 % of small angiolipomas demonstrate some degree of posterior acoustic shadowing [67].
Renal US is indicated for screening purposes in genetic syndromes associated with WT, such as Beckwith–Wiedemann syndrome and Simpson–Golabi–Behmel syndrome. Screening is generally performed every 3 months for the first several years of life, typically at least until the age of 8 [68].
Renal Transplantation in the Pediatric Population
Ultrasound has an excellent track record in the evaluation of renal allograft function and assessment of complications after transplantation. Transplant kidneys are typically placed in the retroperitoneum, generally in the right iliac fossa, with vascular anastomoses to the external iliac arteries. Small pediatric patients do not have adequate space in the retroperitoneum for an adult kidney, and in this situation the kidney is placed intraperitoneally with vascular anastomoses to the aorta and inferior vena cava [69]. Most transplant surgeons obtain an initial baseline ultrasound within 1–2 days postoperatively.
Vascular complications are the most feared in the early postoperative period. Renal artery thrombosis is rare (< 1 %) but devastating. It typically occurs within 1 week and is visualized as absence of flow distal to the renal artery in a hypoechoic kidney. Segmental thrombosis can occur with wedge-shaped areas of the kidney that lack flow on Doppler [70]. Absence of renal vein flow with reversal of arterial flow during diastole is a sign of renal vein thrombosis. Significant vascular compromise in the immediate postoperative period generally requires urgent operative revision. RAS of the transplant artery usually develops within a year of transplant and shows the same signs as in the native kidney, with increased systolic velocity and tardus et parvus waveforms [71].
Ultrasound is very sensitive in the detection of perinephric fluid collections, but cannot reliably distinguish between hematoma, urinoma, and lymphocele. Generally, hematomas appear in the immediate postoperative period, while urine leaks often present after several days and lymphoceles after several weeks [72]. Hematoma tends to have a very heterogenous appearance on ultrasound as opposed to the simple anechoic fluid of lymphocele or urinoma. In the case of ongoing diagnostic uncertainty, aspiration of the fluid may be helpful in determining the etiology as a urine leak demonstrates a very high creatinine level in the aspirated fluid, while a lymphocele has a composition similar to serum. Urine leak from an intraperitoneal kidney leads to ascites . Treatment of a urine leak may include percutaneous nephrostomy or stenting to divert the urine flow and allow for healing of the ureteral anastomosis, but open surgical revision is necessary in large leaks or where ureteral necrosis is suspected. [73] Lymphoceles are frequently septated on US and benefit from image-guided drainage with or without concomitant sclerosis, but operative marsupialization may be necessary if persistent [74].
Measurement of the vascular resistive index (RI) is an essential part of the transplant assessment. It is measured at the arcuate or interlobar arteries and is calculated by the formula (peak systolic velocity—end diastolic velocity)/peak systolic velocity [70]. Less than 0.7 represents a normal value. Elevated RI may be found in acute tubular necrosis, rejection, renal vein thrombosis, and renal dysfunction due to immunosuppressive toxicity from calcineurin inhibitors. Unfortunately, rejection, calcineurin toxicity, and acute tubular necrosis all have nonspecific imaging findings and elevated RI, and renal biopsy is often needed in the case of graft dysfunction without clear vascular abnormalities [9, 75].
Ultrasound Guidance in Renal Biopsy
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


