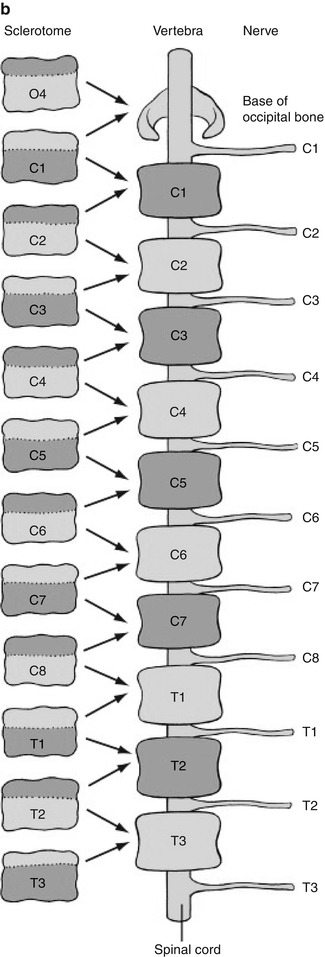
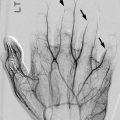
Fig. 3.1
Sclerotome division and reconvergence. Sclerotome division allows the spinal nerves to emerge from the neural tube and extend to the periphery (a). The sclerotome then reconverges to form the final vertebrae (b) (Reprinted with permission from Schoenwolf et al. [127])
The sclerotome grows to surround the notochord and neural tube and then forms the annulus fibrosus. The enveloped notochord goes on to form the infantile and childhood nucleus pulposus. Once adjacent to the notochord and neural tube, the sclerotome divides, allowing the spinal nerves to emerge from the neural tube. When division of the sclerotome is complete, the caudal half of the sclerotome above merges with the cranial half of the sclerotome below to form the vertebra precursor. Fibrocartilagenous cells gradually replace the original notochord cells of the nucleus pulposus.
Chondrification of the membranous vertebrae occurs between fetal weeks 4 and 7. Two cartilaginous centers form in the centrum (either dorsal-ventral or side-by-side). Every neural arch and every costal/transverse process develop one center as well. Ossification begins in the neural arches at fetal weeks 8–9, starting at the upper cervical region. Ossification of the vertebral bodies starts in the lower thoracic and upper lumbar region and moves caudally and cranially; this process is complete by about week 20. All vertebrae below C2 and above the level of the sacrum have three ossification sites: one for the vertebral body and for each neural arch (Fig. 3.2). The single vertebral body ossification center results from coalescence of the two chondral centers that are transiently present (dorsal-ventral or side-by-side), which helps explain the occurrence of posterior and lateral hemivertebrae.
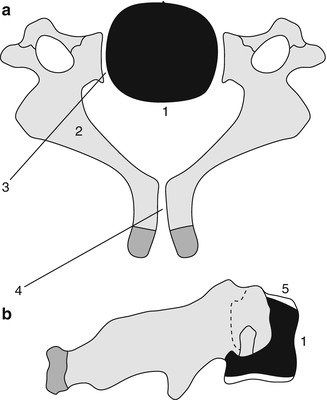
Fig. 3.2
Formation of a lower cervical vertebra. Axial (a) and sagittal (b) views. The body (1) appears by the fifth fetal month. Neural arches (2) appear by the seventh to ninth fetal week and are separated from the body by the neurocentral synchondroses (3), which close between 3 and 7 years of age. The posterior synchondrosis (4) between the spinous processes usually closes by age 2–4 years. Superior and inferior epiphyseal rings (5) appear at puberty, contribute to the vertical growth of the vertebral body, and unite with it at about age 25 years (Reprinted from Ghanem et al. [4] with kind permission from Springer Science and Business Media)
The strip of cartilage that separates the two neural arch centers from the single vertebral centrum, the neurocentral synchondrosis, is slightly anterior to the base of the pedicles (Fig. 3.3). The age at closure of the neurocentral synchondroses varies by position—cervical (3–7 years), lumbar (4–10 years), and thoracic (5 to >10 years) [3, 4]. The ossified portions of each neural arch fuse with each other dorsally by 2–4 years of age; the radiolucent lines seen on the frontal projection of the spine before this age should therefore be recognized as normal (Fig. 3.3). Between 3 and 6 years of age, this synchondrosis disappears as bone bridges the segment, although more distal levels may fuse later [5].
Fig. 3.3
Normal spine synchondroses. (a) Frontal view shows unfused neural arches (arrow) in a 3-month-old infant. (b) Normal neurocentral synchondrosis (arrow) in a neonate, shown on a lateral radiograph. (c) Computed tomography (CT) shows normal unfused neural arch (asterisk) and bilateral unfused neurocentral synchondroses (arrowhead)
Around puberty, secondary ossification centers appear at the tips of the spinous, transverse, and mammillary processes, as well as in the cartilaginous ring apophysis surrounding the vertebral end plate (note that this ossification can appear as early as 2–6 years of age) [6]. Complete rings are rarely visible—on lateral radiographs, they appear as small ossification centers in notches adjacent to the anterior and posterior borders of the vertebral end plates. These secondary ossification centers fuse with the vertebral body in the third decade, usually by age 25.
Ossification of the atlas and axis is somewhat different. Most of the cranial half of the first cervical sclerotome normally merges with the occipital condyles, with a small portion forming the tip of the dens. The caudal half of the first cervical sclerotome forms the anterior, lateral, and posterior masses as well as the posterior arch of the atlas (C1). In the atlas, three separate ossification centers are present in the lateral masses and anterior arch (Fig. 3.4). The anterior arch’s center is ossified at birth in only 20 % of infants and may not be radiographically evident until 2 years of age [7]. Incomplete incorporation or failure of segmentation of the first cervical sclerotome can cause a broad spectrum of fusion anomalies involving the craniocervical junction and accessory structures.
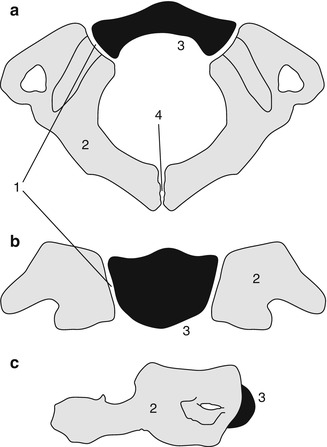
Fig. 3.4
Development of the atlas (C1). Axial (a), coronal (b), and sagittal (c) views. The neurocentral synchondrosis (1) unites the neural arch (2) to the anterior arch (3) of the atlas. The ossification center of the anterior arch (3) appears between age 6 months and 2 years. The posterior synchondrosis at the spinous process (4) unites by 4–6 years of age, followed by the neurocentral synchondrosis (1), which fuses by about age 7 (Reprinted from Ghanem et al. [4] with kind permission from Springer Science and Business Media)
The usual pattern of ossification occurs in the centrum and neural arches of the axis. However, two additional centers form in the base of the dens and fuse into a single odontoid center, either by birth or a few months later (Fig. 3.5) [8, 9]. The ossification centers of the body and dens are separated by a well-demarcated synchondrosis, the homologue of the disc between C1 and C2 (Fig. 3.6). This synchondrosis closes between 3 and 11 years [5] and the neurocentral synchondroses by 3–7 years. A small ossification center appears at the tip of the odontoid between 3 and 10 years; it fuses between 10 and 12 years [5].
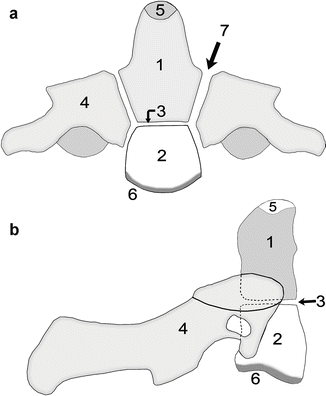
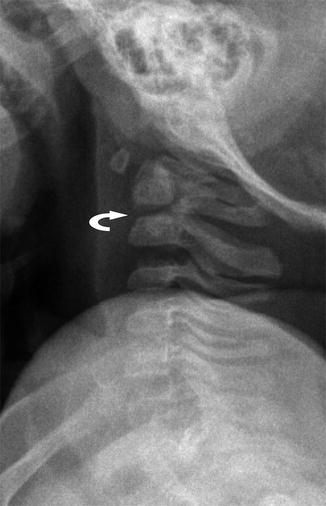
Fig. 3.5
Development of the axis (C2). Coronal (a) and sagittal (b) views. The dens (odontoid process) (1) develops from two primary ossification centers that usually coalesce within the first 3 months of life. The C2 centrum (2) appears by the fifth fetal month and is separated from the dens (1) by the dentocentral synchondrosis (3). The dentocentral synchondrosis fuses between ages 3 and 11, though it usually closes by age 6–7 years. The bilateral neural arches (4) appear by the seventh fetal month and fuse posteriorly by age 3 years. The neurocentral synchondroses (7), located on each side between the dens/vertebral body and neural arch, close between 3 and 7 years. The ossiculum terminale (5) at the tip of the dens appears between 3 and 10 years of age and fuses to the dens between ages 10 and 12. The inferior epiphyseal ring (6) appears at puberty and fuses at about age 25
Fig. 3.6
Dentocentral synchondrosis at the base of the odontoid (curved arrow) in a 3-year-old boy
At the sacrum, in addition to the usual three centers for the body and neural arches of each segment, there are two epiphyseal plates that provide secondary ossification centers to form the superior and inferior portions of the sacral ala of the first through third segments.
Most people have 33 vertebrae: 7 cervical, 12 thoracic, 5 lumbar, 5 sacral, and 4 coccygeal. About 5 % have variant distribution (e.g., 6 lumbar and 11 thoracic vertebrae) but still have a total of 33 vertebral bodies. Three percent have one or two extra vertebrae, and 2 % have only 32 vertebrae [10, 11].
The lumbosacral junction is unstable during embryogenesis, and variations in formation and ossification are common in this area. Transitional vertebrae occur in approximately 20 % and often involve the sacrococcygeal and lumbosacral junctions. These vertebrae retain some features of the adjacent segments and often do not alter the overall number of vertebrae. Common variants include “sacralized” L5 and “lumbarized” S1 vertebrae (Fig. 3.7). Hemi-sacralization or lumbarization can be seen in up to 6.5 % and can cause back pain due to nonuniform articulation. Symptomatic abnormal articulation, known as Bertolotti syndrome, may be characterized on magnetic resonance imaging (MRI) by adjacent edema (Fig. 3.8). Confusion is common when it comes to labeling levels of transitional vertebrae—complete evaluation of the spinal axis before surgical intervention will allow correct assignment of vertebral levels.
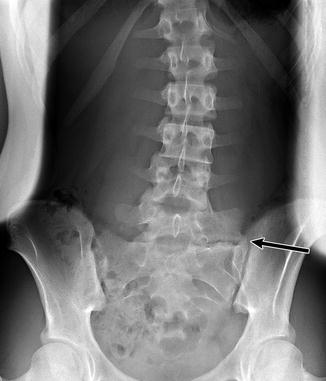
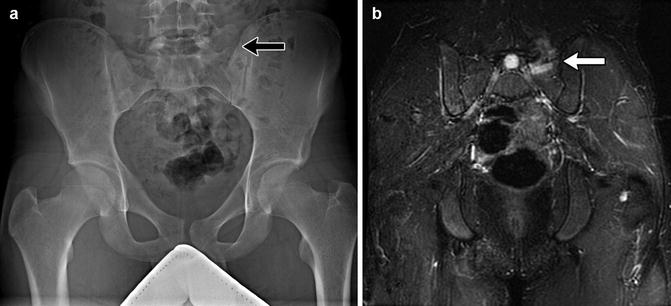
Fig. 3.7
Transitional S1 vertebra in a 14-year-old girl with scoliosis. Note pseudoarticulation of S1 and S2 on the left (arrow) (Courtesy of Thomas Ray Sanchez)
Fig. 3.8
Bertolloti syndrome in a 15-year-old girl with back pain. (a) Transitional vertebral segment with pseudoarticulation on the left (black arrow). (b) Coronal inversion recovery (IR) image shows hyperintensity due to marrow edema on both sides of the pseudoarticulation (white arrow) (Courtesy of Chirag V. Patel)
The individual vertebral bodies enlarge circumferentially by the process of periosteal apposition and grow vertically by endochondral ossification; the cartilage of the vertebral end plates functions like other growth plates. The ring apophyses do not contribute to vertical growth. Following a relatively rapid length increase during the first 2 years of life, the spine grows linearly until age 16 [12]. Normal weight bearing helps balance vertical and horizontal growth and maintains an acceptable ratio between axial diameter and vertebral height. Without normal upright or walking postures, as in children with spinal muscular atrophy or cerebral palsy, vertebral bodies grow disproportionately tall and thin, developing biconvex end plates and decreased disc space height (Fig. 3.9).
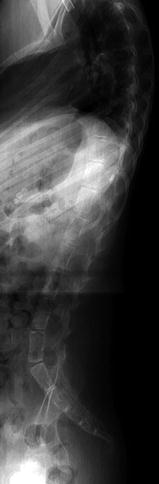
Fig. 3.9
Increased height of vertebral bodies (caninization) in an 11-year-old boy with congenital myopathy
1.2 Variant Ossification
Common developmental features as well as unusual normal variants can be easily mistaken for pathologic processes. The variants described below are common but not exhaustive. Ultimately, consultation with atlases of normal variants when bony defects are uniform, disproportionate to physical exam findings, or asymptomatic should prevent mischaracterization of congenital variants as disease.
The multiple primary and secondary ossification centers and their intervening synchondroses are often mistaken for fractures or avulsion fragments. Ossification in the superior and inferior ring apophysis may begin in children as young as 6 years of age and may persist well into adolescence.
During adolescence, the nucleus pulposus may herniate through the vertebral end plate, resulting in Schmorl nodes (Fig. 3.10). Additionally, notochord remnants may appear as impressions on consecutive inferior end plates, mimicking erosive or lytic lesions. Similarly, anterior intraosseous herniation of the intervertebral disc, also referred to as a limbus vertebra (Fig. 3.11), may be mistaken for a teardrop fracture. These normal variants may be differentiated from fractures by examining the edge of the lesion: physeal plates should be smooth and regular, with subchondral sclerotic lines, whereas acute fractures are irregular and non-sclerotic; fractures can occur at any location.
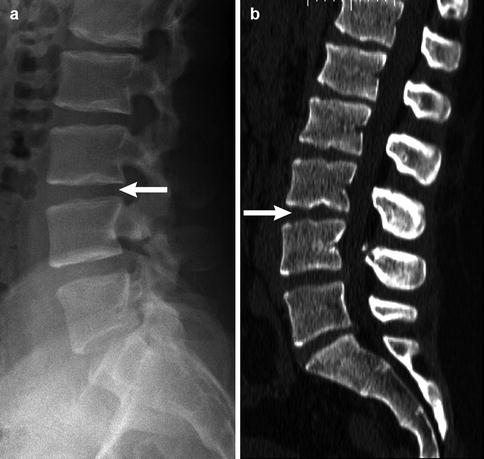
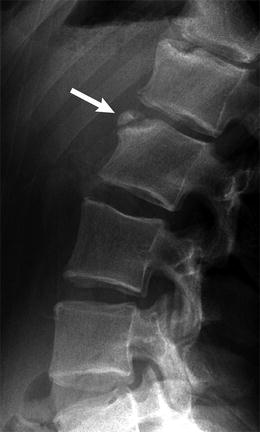
Fig. 3.10
Multiple Schmorl nodes in a 17-year-old male. Arrows delineate L3–4 on radiograph (a) and sagitally reformatted CT (b) (Courtesy of John Hunter)
Fig. 3.11
Limbus vertebra at L2 (arrow) in a 16-year-old girl
During the first two decades, the appearance and physiology of the cervical spine varies tremendously. Until age 15, a normal atlantoaxial interval can be as large as 5 mm and occasionally even 6 mm. More accurate assessment evaluates for change in distance between passive flexion and extension, which should not exceed 2 mm.
Physiologic displacement of C2 anteriorly on C3 is common in normal children due to ligamentous laxity (Box 3.1). Known as pseudosubluxation, this occurs because until age 10, flexion and extension are centered at the C2–3 level (Figs. 3.12 and 3.13). (In adolescence and throughout adulthood, maximum mobility is at C4–6.) In order to determine whether subluxation results from this normal variant or from a C2 arch fracture and pathologic subluxation, one can draw a curved “posterior cervical” line, connecting the anterior cortices of the C1 and C3 spinous processes (Fig. 3.12). With physiologic subluxation, this line either passes through the C2 posterior arch or passes less than 2 mm anterior to it. In the presence of an arch fracture, the distance will be greater, since the atlas and the C2 centrum slip forward at the fracture site while the spinous process remains posterior [13, 14].
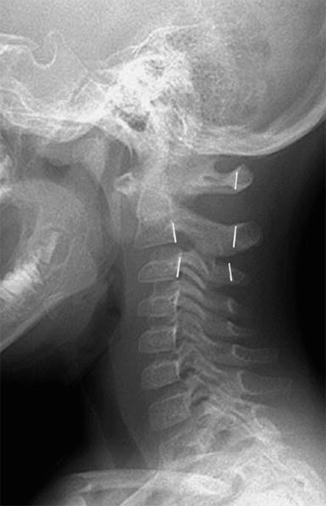
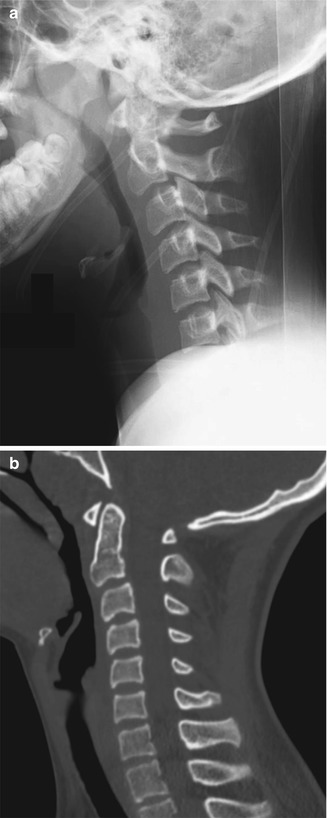
Fig. 3.12
Normal pseudosubluxation of C2 on C3. A discontinuous line connects the posterior C2 and C3 vertebral bodies, but the posterior cervical line along C1–3 is continuous and normal (Reprinted from Cakmakci [14] with kind permission from Springer Science and Business Media)
Fig. 3.13
Normal pseudosubluxation of C2 on C3. Radiograph (a) and sagitally reformatted CT (b) show forward displacement of C2 on C3. However, the posterior cervical line is normal
Box 3.1: Pseudosubluxation of C2–3
Before about age 10 years |
Due to normal ligamentous laxity |
Posterior cervical line connects the anterior cortex of spinous processes of C1 and C3 |
Passes through the posterior arch of C2 or <2 mm anterior to it |
Developmental pseudospread of the atlas is a normal phenomenon that occurs because the growth of the atlas is linked to the relatively rapid growth of the skull base, whereas the growth of the axis is influenced by the slower growth of the spine. With pseudospread, the lateral margins of C1 lie symmetrically lateral to the margins of C2, mimicking the bilateral lateral displacement of a Jefferson fracture of the arch of the atlas. This disparity generally resolves after age 2. The overlap is usually less than 2 mm, whereas in the case of fracture, it generally exceeds 3 mm [15].
In up to 5 % of newborns, a coronal cleft is apparent on a lateral radiograph of the thoracic or lumbar spine. This appears as a vertical lucency at the junction of the anterior two thirds and posterior third of the vertebral body [16]. The cleft is filled with hyaline cartilage, possibly a remnant of the division between dorsal and ventral ossification centers. The coronal cleft ossifies during infancy and does not persist or lead to other anomalies.
A horizontal cleft in the midportion of the vertebral body, also referred to as a Hahn cleft, may also be visible on a lateral view. It marks both the boundary of fusion between sclerotomes and the nutrient foramen (Fig. 3.14) and may persist as a sclerotic line throughout adolescence.
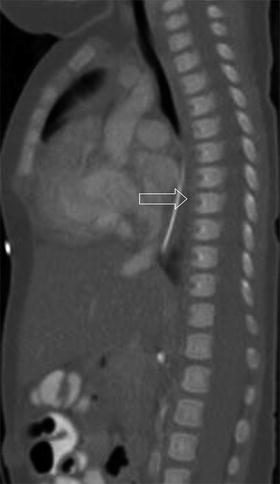
Fig. 3.14
Hahn clefts (arrow), the boundary of fusion of the reformed sclerotomes and also the level of arterial entry into the vertebral bodies (sagittally reformatted CT)
Bone-in-bone is another common finding during infancy (Fig. 3.15). This results from rapid growth during the first postnatal months, before remodeling of the fetal spine begins [14]. The outline of a smaller replica of the vertebral body is seen within the centrum.
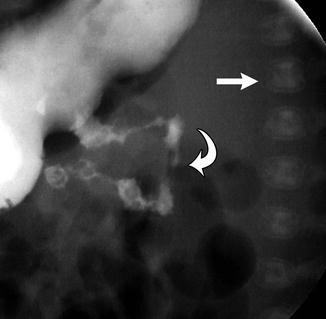
Fig. 3.15
Bone-in-bone (arrow) in a newborn with malrotation (curved arrow) (Lateral view from a fluoroscopic study)
Incomplete bony fusion at the neural arch of L5 or S1 is very common in children, but the incidence decreases during childhood. Cartilage usually fills this apparent gap, so it is not functionally open [17]. The S1 neural arch is incompletely fused in up to 58 % at age 8, 48 % at age 12, and 10–26 % of adults. A separation of 3 mm or less is normal and will often ossify during childhood. Occasionally, a similar narrow cleft may be encountered as an isolated finding at the T11 or T12 level. The term spina bifida occulta has been applied to this appearance (at any isolated level), but this term may be misleading as it implies pathology. Misreporting of this finding may result in confusion and unnecessary imaging.
At the T12 and L1 vertebrae, localized moderate to marked pedicle thinning occurs in 7 % of normal children and adults. The developmentally aberrant pedicle may be differentiated from a pedicle that is being deformed by mass effect because the lateral border of the aberrant pedicle is flat or concave, rather than rounded.
2 Structural Vertebral Abnormalities
Spinal defects may be restricted to the vertebrae or may involve neural and non-neural structures [18]. Sixty-one percent of those with congenital spinal defects have other abnormalities as well. For example, a prenatal neural crest and sensory nerve injury may lead to a field defect that causes the constellation of upper limb, vertebral, and rib anomalies. The mnemonic VACTERL describes the clinical confluence of vertebral, anorectal, cardiac, tracheoesophageal, renal, and limb anomalies [19]. It is important to recognize that VACTERL is an association not a syndrome. Less common associated abnormalities are found in the ear, choanae, lips, palate, intestinal tract, abdominal wall (hernias), and genital system.
Scoliosis may develop if vertebrae fail to form, as with wedge vertebrae (unilateral hypoplasia) or hemivertebrae (unilateral aplasia). Less often, a hemivertebra may be supernumerary, resulting from partial duplication. Failure of segmentation may be unilateral or bilateral, resulting in an osseous bar (or bars) connecting the posterior elements, vertebral bodies, or both (Fig. 3.16). If anomalies are multiple and balanced, alignment may be normal.
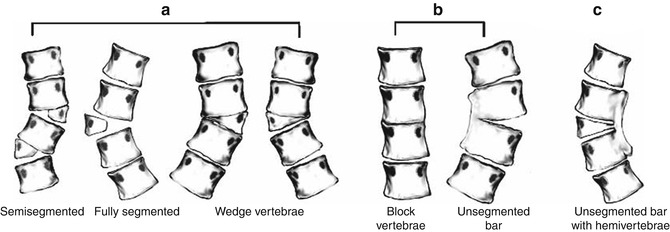
Fig. 3.16
Diagrammatic representation of the classification system of congenital scoliosis. (a) Failures of formation: various types of hemivertebrae (semisegmented, fully segmented, wedge vertebra). (b) Failures of segmentation: block vertebra, unsegmented bar. (c) Mixed: combination of hemivertebra and unsegmented bar (Reprinted with permission from Erol et al. [128])
2.1 Vertebral Anomalies
Wedge vertebrae (Fig. 3.16) form asymmetrically such that one side is hypoplastic and the other develops normally. Both pedicles are present, but one is larger than the other. A single wedge vertebra results in scoliosis, with the apex at the abnormal vertebra. However, bilateral wedge vertebrae may balance each other and result in a straight spine [20].
Hemivertebrae (Figs. 3.16, 3.17, and 3.18) are completely aplastic on one side, resulting in a single unpaired pedicle attached to a triangular vertebral half-body [21]. Hemivertebrae usually replace a normal vertebra but if supernumerary tend to be positioned slightly more laterally than are the other vertebrae. The hemivertebra may fuse with an adjacent vertebral body (“nonsegmented”), creating a tripediculate block vertebra with two pedicles on one side but only one on the other. If the hemivertebra is not fused to adjacent vertebrae (“fully segmented”), it is more likely to cause continued asymmetrical growth [21], since it has two functioning—and tilted—growth plates. However, curve progression is less likely with a tripediculate block vertebra. The hemivertebra may also be semisegmented, in which case one growth plate is fused to the adjacent vertebra and the other is not. Computed tomography (CT) or magnetic resonance imaging (MRI) helps assess the extent to which a hemivertebra is fused to the adjacent vertebrae [22].
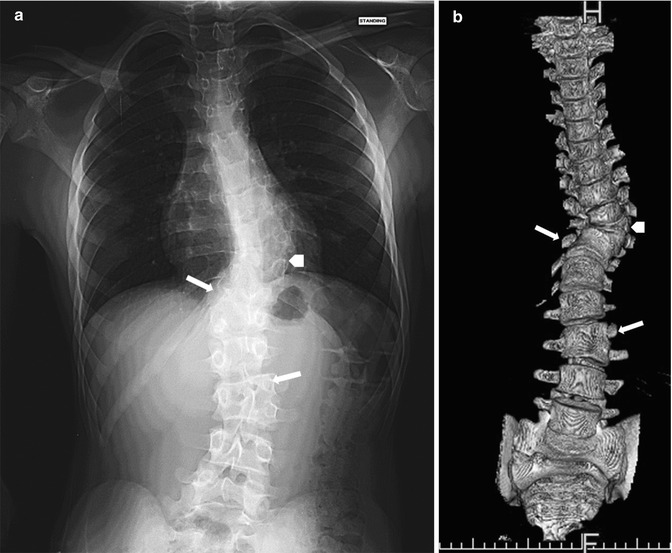
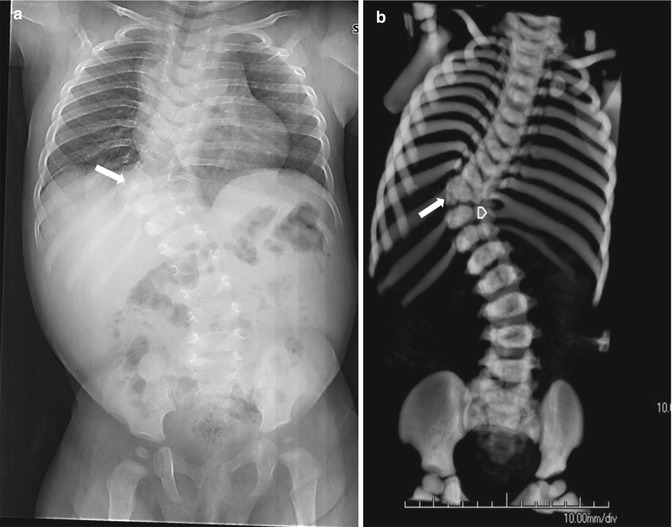
Fig. 3.17
Congenital scoliosis. Radiograph (a) and 3-dimensional (3D)-CT reconstruction (b) show three hemivertebrae on the left. The most cephalad (arrowhead) is fully segmented, whereas the more caudad are semisegmented (arrows), fused to the vertebra below
Fig. 3.18
Congenital rotatory scoliosis. Radiograph (a) and 3-dimensional (3D)-CT reconstruction (b) demonstrate a right-sided hemivertebra at T9–T10 interspace (arrow). Even though the ribs end at same level, the right side has 12 ribs and the left has 11, because of the extra rib induced by the right hemivertebra. Also note fusion of the left 9th and 10th ribs (arrowhead)
The pedicle of the hemivertebra may induce rib development. Additionally, ribs at the most concave portion of the scoliosis may fuse. Counting all the ribs on both sides is an essential step in assessing the configuration and significance of wedge vertebrae and hemivertebrae. Since hemivertebrae may be supernumerary, accurate assessment of vertebral level requires evaluation of the entire spine, from C1 through the coccyx.
Butterfly vertebrae (Fig. 3.19) have a partial sagittal cleft resulting in two approximately symmetric segments that can be otherwise normal or appear as bilateral wedge vertebrae or—if completely divided—hemivertebrae. If balanced, butterfly vertebrae do not cause significant scoliosis, but they may suggest the presence of a syndrome or other anomalies.
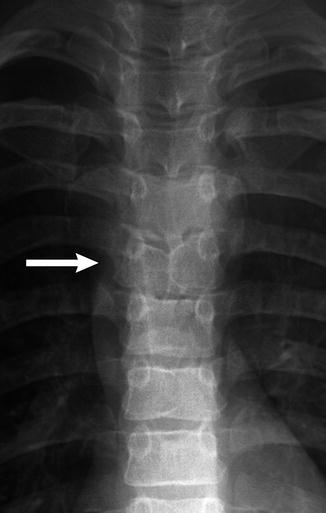
Fig. 3.19
Butterfly vertebra (arrow) in an 11-year-old boy with Alagille syndrome
Isolated absence of the pedicle (Fig. 3.20) is most common in the cervical and lumbar region and is uncommon in the thoracic spine. It is important to distinguish this congenital anomaly from pathologic destruction of the pedicle. With congenital absence of the pedicle, the pedicle outline is completely absent, and the associated intervertebral foramen is enlarged. In older children, the opposite pedicle and lamina may be hypertrophied.
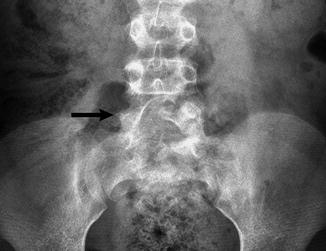
Fig. 3.20
Transitional lumbosacral vertebra in a 6-year-old girl. The pedicle on the right (arrow) is absent
Congenital vertebral bars (Fig. 3.21) develop when the laminae or pediculate bases of adjacent vertebrae fuse. Fusion that is limited to the laminae and facets dorsal to the intervertebral foramina leads to lordo-scoliosis. If the vertebral bodies at the base of the pedicles are also fused, the intervertebral foramina will be small. If there are no ribs in this region, a large conglomerate foramen develops, through which multiple nerve roots pass. If these anomalies are bilateral and symmetric, growth will be restricted but alignment may be normal. If unilateral, more problematic angular deformity occurs.
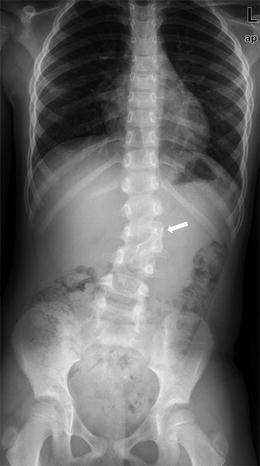
Fig. 3.21
Laminopediculate bar. The lamina of the hemivertebra has fused to the lamina of the pedicle above (arrow)
If cleavage of somites fails, normal segmentation does not occur, and block vertebrae form. Vertebral bodies do not separate, and there is no (or very little) intervening disc space. The vertebral block appears constricted at the level of the disc, and the height of the structure is slightly less than that of two normal vertebral bodies (excluding the height of a disc). A thin sclerotic linear density may replace the disc, or a rudimentary disc may be present.
Fusion in young children is often cartilaginous and thus may be radiographically occult. In older children, a frontal radiograph demonstrates a laminopediculate bar as a discrete sclerotic curved line connecting pedicles at two or more levels (see Fig. 3.21). The ipsilateral vertebral body and disc space are often hypoplastic, and the vertebral bodies may be fused as well. If the fusion is coronal rather than sagittal, the anterior aspect of the vertebral body is almost invariably wedge shaped or entirely absent, resulting in kyphosis or lordosis (further discussed below). In young children, radiographs may fail to demonstrate fusion, but MRI readily depicts block vertebra formation by demonstrating absence of intervertebral disc signal (Fig. 3.22).
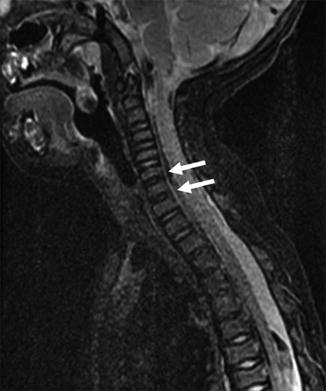
Fig. 3.22
Lack of normal hyperintense intervertebral disc signal in the upper thoracic and lower cervical region (arrows) denotes areas that will develop into block vertebrae when ossification is complete (sagittal T2-weighted (T2-W) fat-suppressed (FS) magnetic resonance image (MRI)) (Incidental flow artifact dorsal to the spinal cord)
CT is valuable in showing associated spinous process obliquity, transverse process hypoplasia, abnormally shaped spinal canal, anomalous facet and rib articulations, and hypoplasia of pedicles at adjacent levels. It also delineates hypoplastic or malpositioned pedicles that are not apparent on routine radiography. MRI best images nerve root compression, myelomalacia, and contralateral laminar stress fracture, all of which can cause back pain.
2.2 Congenital Scoliosis (Table 3.1)
I. Failure of formation |
A. Unilateral hypoplasia (wedge vertebra) |
B. Unilateral aplasia (hemivertebra) |
II. Partial duplication (supernumerary hemivertebra) |
III. Failure of segmentation |
A. Unilateral nonsegmentation (unilateral bar) |
1. Posterior elements only |
2. Vertebral body only |
3. Vertebral body and posterior elements |
IV. Combined lesions |
Spinal curvature in the coronal plane is caused by unbalanced anatomy and growth potential of the two sides of the spine. Unlike idiopathic scoliosis, congenital scoliosis progresses in up to 75 % of patients and most often affects the thoracic spine (thoracolumbar scoliosis is the second most common). Once curvature starts to increase, it will continue to do so, often accelerating during the preadolescent growth spurt. Since ribs and vertebrae form from the same mesenchymal somites, associated developmental and fusion anomalies are common. However, it is difficult to predict exactly what effect a single anomaly will have.
Multiple lesions are sometimes balanced, canceling each other out and causing little overall effect on spinal alignment. Ribs that fuse centrally near the spine may contribute to scoliosis, but peripheral rib fusion generally does not affect spinal curvature [23].
Hemivertebrae function both as the apex of the curve and as an unbalanced growth center that contributes to increased curvature. Congenital bars have the opposite effect of hemivertebrae, restricting growth on one side of the spine while the other side continues to grow. Congenital bars are present in 40 % of patients with multiple hemivertebrae. A hemivertebra at the same level as a contralateral bar has the worst prognosis, especially if thoracolumbar. However, in young children it can be difficult to predict exactly where and how a segment will fuse to adjacent bodies.
Imaging
Radiographic evaluation includes frontal and lateral radiographs. Curves can be measured according to a variety of techniques; the Cobb technique is commonly employed and is presented later in this chapter. Cross-sectional imaging is indicated for exact delineation of anomalies [24]. CT and MRI may demonstrate occult hemivertebrae as well as vertebral fusion. Additionally, MRI of the entire spine is typically performed before surgery in order to assess asymptomatic associated anomalies such as tethered cord, syringomyelia, and diastematomyelia, which are cumulatively described in up to 40 % of patients with congenital scoliosis [25].
Up to 18 % of patients with congenital scoliosis have associated genitourinary tract abnormalities, with renal agenesis being the most common [26]. Urologic evaluation is therefore indicated in all patients with congenital anomalies of the spine. Screening ultrasound (US) to assure the normal site and structure of both kidneys usually suffices, although occasionally additional imaging is necessary. Patients may also have congenital heart disease (7 % incidence), undescended scapula (6 %), thumb anomalies (4 %), and other malformations.
2.3 Congenital Kyphosis and Lordosis
Congenital kyphosis is another manifestation of unbalanced longitudinal development of vertebrae, with the curvature occurring along the sagittal axis. As with coronal scoliosis, these developmental anomalies generally manifest as failure of formation, failure of segmentation, or both.
Kyphosis in the thoracic spine is defined as curvature greater than the 40° [27–29]. It can be caused by posterior overgrowth, restricted anterior growth, or anomalies of segmentation [30]. The most common form of congenital kyphosis is caused by a dorsal hemivertebra (although infrequently the entire vertebral body is absent but the neural arch elements are formed) (Fig. 3.23). Both the anterior and the lateral portions of the vertebra may be deficient, causing a combination of scoliosis and kyphosis [31]. If kyphosis is severe, an anterior pseudoarticulation may develop above or below the defect. Anterior block vertebra formation rarely causes kyphosis, usually without scoliosis, since such fusion tends to be symmetric [32].
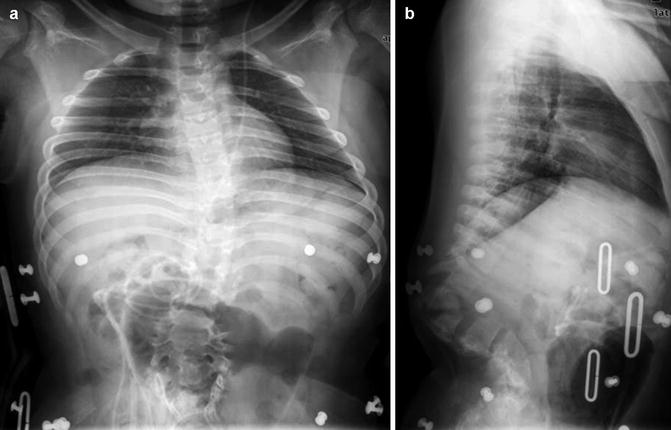
Fig. 3.23
Congenital thoracolumbar kyphosis and posterior spinal dysraphism. (a-frontal, b-lateral) Multiple anterior vertebral body segments are hypoplastic, and posterior elements have not fused. The patient is in a brace, and there is a ventriculoperitoneal shunt catheter
Congenital nonsegmentation and fusion must be distinguished from progressive anterior vertebral body fusion, in which erosion of the anterior end plates is followed by anterior fusion and later disc fusion. Muscle imbalance in severe myelomeningocele may also cause kyphosis.
Congenital lordosis, a lumbar angle over 40°, is usually severe but fortunately uncommon. In this condition, posterior elements fuse while the anterior vertebral bodies continue to grow. Extreme thoracolumbar lordosis may also result from intrauterine positioning. Posterior rib synostosis contributes to growth inhibition.
Kyphosis and lordosis are usually assessed with standing radiographs, with flexibility determined by placing the patient on a bolster. As with congenital scoliosis, CT clearly delineates osseous causes of kyphosis or lordosis. MRI best evaluates for spinal cord compression and other cord abnormalities. A complete investigation includes cardiac and urologic screening due to the high associated incidence of these anomalies.
2.4 Craniovertebral Junction
Like the lumbosacral area, the craniovertebral junction is developmentally unstable. Congenital anomalies and developmental variants in this region are therefore common. Anomalous development here often results in multiple abnormalities, and therefore recognition of a single variant should trigger evaluation for additional problems. Many patients with abnormal development at the craniocervical junction can further be classified as syndromic [33]. While many abnormalities at the craniocervical junction are minor and asymptomatic, clinically significant spinal cord compression can result from somatic growth or recurrent motion.
Occipitalization of the Atlas
Occipitalization of the atlas (Fig. 3.24) occurs in up to 0.75 % of the population and ranges from complete osseous fusion between the atlas and occiput to more focal osseous bridging. Most commonly, the anterior arch of the atlas fuses to the skull base. However, fusion may involve the posterior arch or the lateral masses, and multiple sites of fusion are not uncommon [33]. Atlantoaxial instability is common in those with occipitalization of the atlas, as is encroachment on the spinal cord, sometimes resulting in myelopathy. Since development of the neural elements is closely intertwined with that of bony elements, basilar invagination, Chiari 1 malformation, and tonsillar ectopia may be present as well. Fifty-seven percent of patients with occipitalization of the atlas also have a syndrome [33].
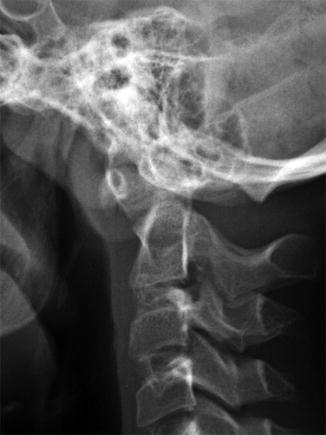
Fig. 3.24
Occipitalization of atlas. There is continuity of the cortex between the skull base and C1 (Same patient as Fig. 3.27)
Occipitalization of the atlas must be distinguished from an unusually high atlas, which can occur as a normal variant due to relatively flat occipital condyles. In some cases, occipitalization is associated with a short clivus, the latter due to primary growth failure (as in achondroplasia) or premature fusion of the spheno-occipital synchondrosis [4].
The posterior arch at C1 may be open in some patients with craniocervical fusion [33], but this finding can be seen until age 4 years in normal children and indeed persists as a normal variant in up to 4 % of adults. Complete absence of the posterior arch of the atlas is rare (Fig. 3.25). Defects in the anterior arch of C1 are less common than defects in the posterior arch. The anterior arch of the atlas rarely fuses to the dens. The occipital condyles may be asymmetrical, and similar asymmetry may be seen at the atlantal and axial articulations. One third of patients have associated fusion of C2–3 vertebrae.
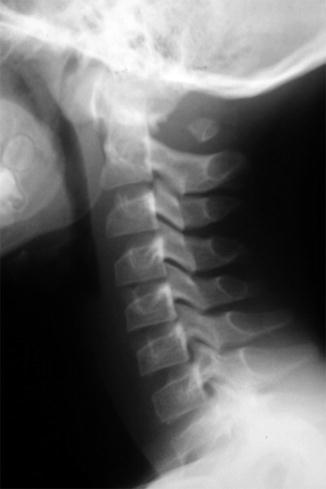
Fig. 3.25
Near complete absence of the posterior arch of C1 in a 12-year-old girl
Patients with craniocervical junction abnormalities may complain of neck pain and stiffness, with neurological signs and symptoms usually beginning insidiously in the third decade and progressing slowly. However, earlier onset is being reported with increasing frequency. Sometimes symptoms occur acutely, and sudden death may result from relatively mild trauma at the craniocervical junction.
Imaging
Fusion may be apparent on a lateral radiograph, showing a fused block of bone or continuous cortex. Lack of mobility on flexion and extension radiographs can indicate fusion as well. This finding is especially valuable in skeletally immature children. However, if there is reasonable suspicion for fusion, CT with sagittal and coronal reformats as well as three-dimensional (3D) reconstructions is most useful to delineate bony anatomy, and MRI may be useful to evaluate cord impingement or myelopathy.
C2 Developmental Anomalies
Multiple anomalies can affect the dens. Some have significant clinical implications, whereas others may contribute to diagnostic uncertainty in the setting of trauma. The dens may be hypoplastic or hypertrophied, or it may be composed of ossicles that fail to fuse to each other or to C2.
The normal odontoid terminates just below the foramen magnum, with a normal basion to (top of) dens measurement of 8.3–10 mm [34]. If the odontoid extends above or below this level, neurologic symptoms may result, especially if there is also transverse ligament laxity [35]. In odontoid hypoplasia, also known as stubby dens, the odontoid process is underdeveloped and does not reach its normal height at the superior surface of the atlas. In the severe form, odontoid aplasia, the dens does not extend above the body of C2. These congenital anomalies of formation may be isolated or associated with syndromes such as mucopolysaccharidoses, spondyloepiphyseal dysplasia, and metatropic dwarfism.
Anomalous development of the odontoid can take several forms. Os odontoideum describes a small fragment of bone that does not fuse with the subjacent portion of the odontoid. The ossific fragment usually lies directly behind the anterior arch of the atlas, but it is sometimes displaced (Fig. 3.26). Occasionally, there is a transverse cleft between the odontoid and the adjacent C2 vertebral body. A history of neck trauma is common in patients with this finding, and the phenomenon may sometimes result from an incomplete or partially healed fracture rather than aberrant development. The unfused ossicle usually moves with the atlas and occasionally fuses to the anterior lip of the foramen magnum.
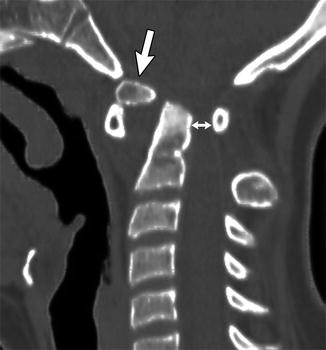
Fig. 3.26
Os odontoideum in an 11-year-old girl with Down syndrome. Sagittally reformatted CT shows the os (arrow) is displaced, positioned just behind and superior to the anterior arch of C1, with the remainder of the dens posterior to the os. There is marked narrowing of the spinal canal (double arrow)
Differentiating os odontoideum from acute fracture can be difficult [36]. Features that suggest a chronic process such as os odontoideum include a relatively thick anterior arch of C1 with a convex posterior border. MRI can also help differentiate by demonstrating normal signal intensity in the bone and surrounding soft tissues.
Atlantoaxial Subluxation
In atlantoaxial subluxation, the transverse, alar, and other ligaments binding the odontoid to the atlas are lax or absent whereas bones are normal. Laxity of the transverse and other stabilizing ligaments allows the anterior arch of the atlas to subluxate anteriorly [37] (Box 3.2). Atlantoaxial subluxation may be associated with structural abnormalities. Alternatively, it may be a component of various syndromes, such as Ehlers-Danlos syndrome and other connective tissue disorders or trisomy 21. The condition can also follow trauma or be associated with local osteomyelitis, pharyngitis and cervical adenitis, and juvenile idiopathic arthritis. Rotational and atlanto-occipital instability as well as skeletal anomalies may be present, and scoliosis occurs in up to 50 % of cases.
Imaging
In normal older children and adults, the gap between the posterior surface of the anterior arch of the atlas and the anterior surface of the odontoid is 2–3 mm, whereas in younger children the presence of cartilage may cause this distance to be as large as 5 mm or occasionally even 6 or 7 mm. Although slight laxity and instability can be present in normal children, the atlanto-dens interval usually does not change with flexion or extension. In atlantoaxial subluxation, the distance is usually normal on standard neutral lateral radiographs but increases 2 mm or more with flexion (Fig. 3.27). However, in some patients with trisomy 21, the atlanto-dens interval is abnormal on neutral images yet normal with flexion [37].
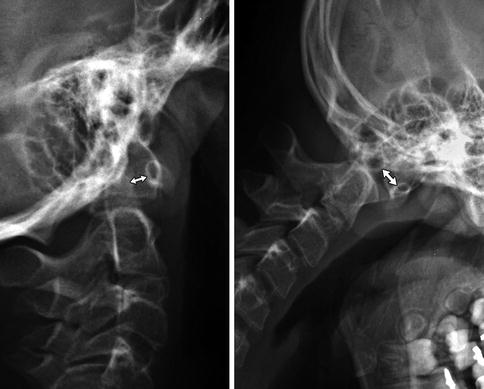
Fig. 3.27
Abnormal motion between the anterior arch of C1 and the dens (double arrows) in patient with occipitalization of the atlas (Same patient as Fig. 3.24)
Box 3.2: True Atlantoaxial Subluxation
Associated with structural abnormalities, trisomy 21 and other syndromes, arthritis, trauma, and pharyngitis |
Increased distance between the anterior arch of C1 and dens |
Normal <5 mm in young children |
Normal <2–3 mm in older children |
Increases with flexion |
Atlantoaxial Rotary Subluxation
Atlantoaxial rotary subluxation may follow trauma or occur with Grisel syndrome (local inflammation in the pharynx) [38]. In the latter condition, inflammation around the dens and pharyngo-vertebral venous plexus causes the ligaments to weaken [39]. Relatively minor trauma to this area that would not affect an adult may lead to rotary subluxation in children, perhaps due to more steeply angled facets.
Imaging
The atlantoaxial facets and occiput-C1-C2 articulation are best investigated with CT as the patient passively rotates their head. Sagittal and coronal reformatted images or 3D-CT reconstructions demonstrate the abnormal relationships well (Fig. 3.28). On fluid-sensitive MRI sequences, abnormal signal in the transverse and alar ligaments is sometimes evident; this may be reactive or causative.
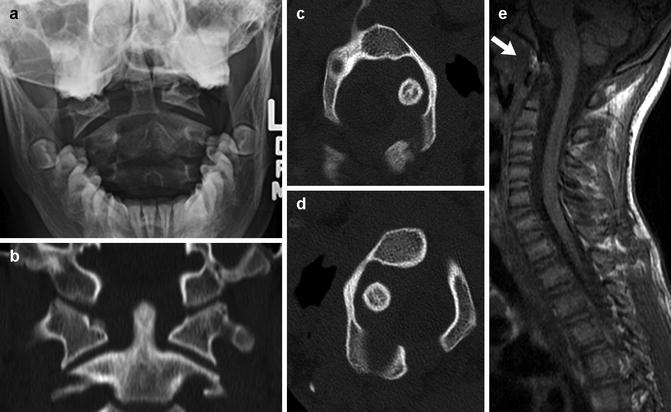
Fig. 3.28
Atlantoaxial rotary fixation in a preteen. (a) Frontal radiograph shows asymmetry between the lateral masses of C1 and the dens, with the head tilted to the right. (b) Coronally reformatted CT in neutral confirms this finding. Axial CT images with the head turned to the right (c) and left (d) show asymmetrical widening on the right persists. (e) Sagittal T1-weighted (T1-W) MRI shows subacute hemorrhage at the alar ligament (arrow)
3 Spinal Dysraphism Complexes
Dysraphism describes the failure of midline vertebral structures to fuse. The term encompasses not only structural vertebral defects but also associated anomalies of neurologic, gastrointestinal, genitourinary, and other systems. The sequence of neural tube-notochord-yolk sac differentiation is disrupted, and thus structures that develop from the primordial layers may be affected. There may be abnormal induction of structures that develop later as well. Dysraphism is categorized as open (uncovered) or closed (covered) and the later further subdivided according to configuration (Fig. 3.29) [40]. Most open defects (meningocele (Fig. 3.30), meningomyelocele (Fig. 3.31), and myelocele) are clinically obvious, but dermal sinuses may be difficult to detect. Closed defects are completely covered with skin, which may be dysplastic and have an associated nevus, hairy patch, hemangioma, or lipoma. The spinal cord may be duplicated or split (diplomyelia and diastematomyelia), or there may be overgrowth of normal tissues (lipoma and dermoid) or herniation of one germ layer through another (neurenteric cyst).
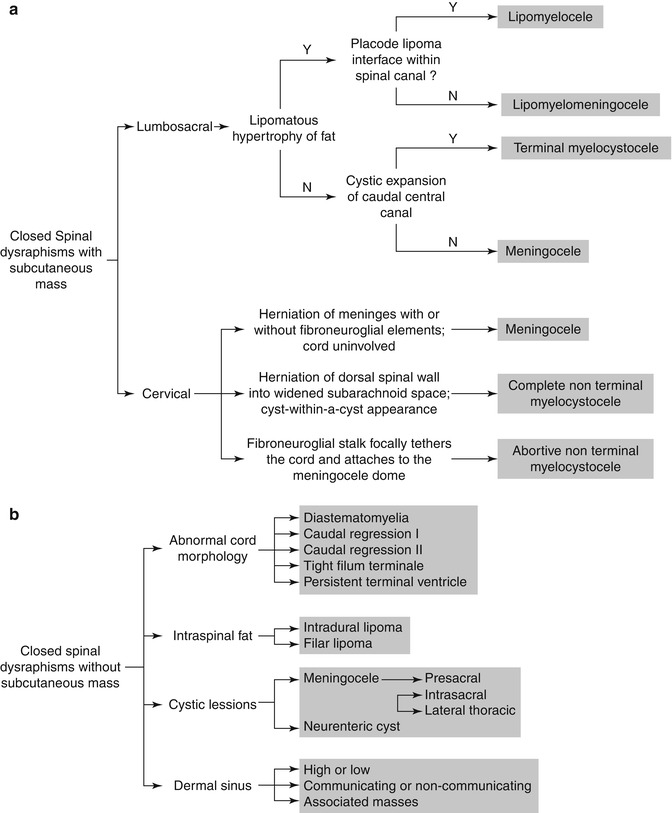
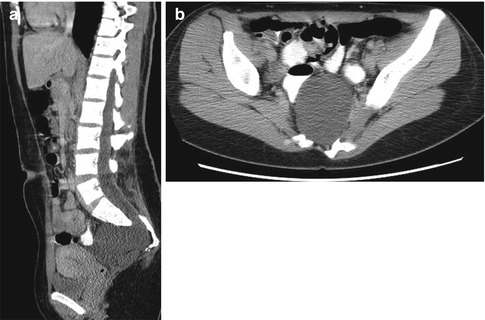
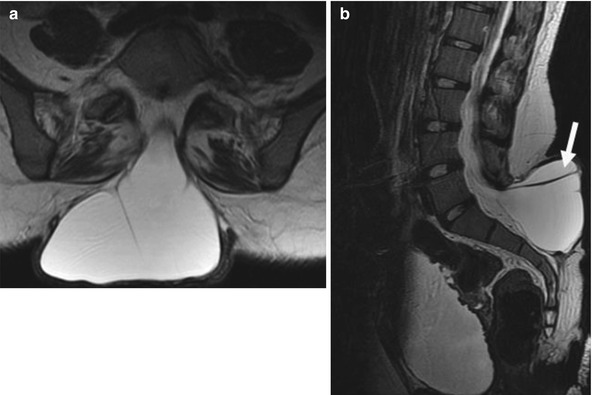
Fig. 3.29
Diagnostic algorithm of closed spinal dysraphism presenting with (a) and without (b) a subcutaneous mass (Reprinted from Badve et al. [129] with kind permission from Springer Science and Business Media)
Fig. 3.30
Anterior pelvic meningocele. Sagittally reformatted (a) and axial CT (b) images show an anterior pelvic meningocele at the mid-sacral level. Dorsal elements are dysmorphic, and there is lower lumbar and sacral dysraphism with partial sacral agenesis
Fig. 3.31
Meningomyelocele. Axial (a) and sagittal (b) T2-W images demonstrate a myelomeningocele at the L5–S1 level with posterior spinal dysraphism and extension of the neural placode into the dorsal aspect of the cyst (arrow). Note the trabeculated neurogenic bladder
Imaging
In both open and closed lesions, radiographs reveal structural vertebral defects in most patients. Neural arches appear widely spread, and laminar/pediculate fusion or intersegmental fusion is common, often affecting more than one level. The spinal canal may appear diffusely enlarged. Vertebral bodies are often hypoplastic, deformed, or scalloped, and there may be hemivertebrae (see Fig. 3.23). These findings may be present at the level of the neurologic defect or elsewhere in the spine. Associated mediastinal or pelvic masses may result from ectopic neurenteric rests.
Cross-sectional imaging provides essential information for skin-covered lesions. If cutaneous stigmata are present, US can help evaluate for tethered cord until about age 3 months (Figs. 3.32 and 3.33), with subsequent MRI if needed. CT is rarely performed to better delineate the bony abnormalities, while MRI exquisitely depicts the extent of neural and soft tissue abnormalities (see Figs. 3.30 and 3.31). MRI may occasionally be performed before surgery for open lesions, as it allows more precise diagnosis and surgical planning.
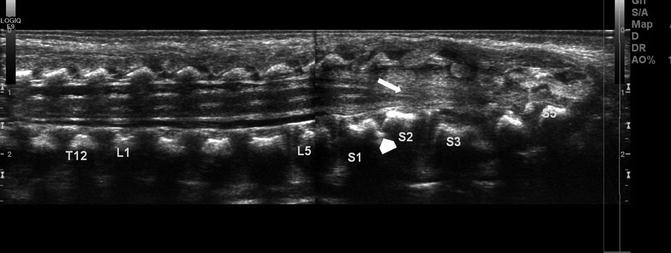
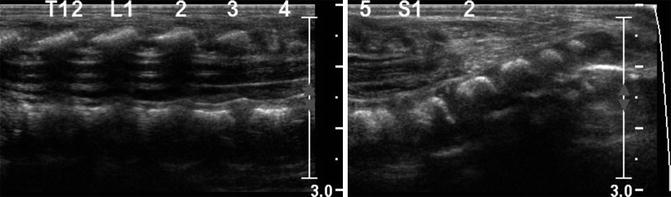
Fig. 3.32
Tethered cord in a newborn. Longitudinal ultrasound (US) shows the spinal cord extends to the sacral-level dysraphism (arrow). Additionally there is a lipoma of the filum. Note the fused vertebrae compatible with VACTERL association (arrowhead)
Fig. 3.33
Tethered cord. Longitudinal US shows conus at the S1 level, with increased fibro-fatty tissue in the canal below this level
3.1 Tethered Filum Terminale
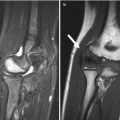
Low conus or tethered cord generally occurs because either the terminal cord fails to involute or the filum terminale fails to lengthen. Low conus (Fig. 3.33) is characterized by abnormal termination of the cord, whereas abnormal thickening of the filum, fibrolipoma, and/or a filar cyst characterize an abnormal filum terminale (Fig. 3.34). If the conus is abnormally low, nerve roots emerge more horizontally than usual, and stretching of the vascular supply may lead to myelopathy. Tethering occasionally presents late with such symptoms as lower extremity spasticity, bladder incontinence, and toe walking. Hairy tufts, midline vascular malformations, and other cutaneous findings should prompt evaluation.
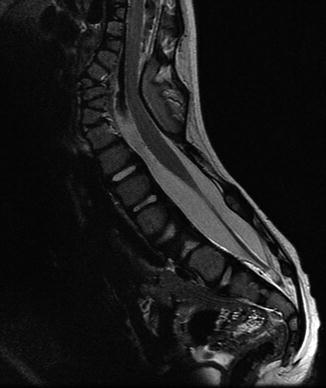
Fig. 3.34
Tethered cord and multiple segmentation anomalies in a 3-year-old. Sagittal T2-W image shows the distal cord is dorsally positioned and kinked over dysmorphic posterior elements at the thoracolumbar junction. Multiple dorsal and ventral hemivertebrae
A conus terminating at the superior half of the L3 vertebral body can be considered normal in newborns, provided that normal ascent follows [41]. The inferior tip of the conus should lie at the adult level, T12 to L2/3, by 2 months of age. Absence of normal motion with cerebrospinal fluid (CSF) pulsation is another feature of tethered cord [42]. Even if surgery reveals that the conus is not tethered, symptoms sometimes occur. Tethering can also occur or recur after surgical repair of myelomeningocele, diastematomyelia, and other lesions.
Imaging
Ultrasound (US), performed between birth and 3 months of age, readily evaluates the level of the conus. Additionally, evaluation of cerebrospinal fluid pulsations can identify abnormal cord motility. If US identifies tethered cord, MRI of the entire neural axis should be performed to evaluate for occult concomitant issues. The filum is abnormally thick if it measures more than 2 mm in diameter at L5–S1 or if it is infiltrated by fibrous or fatty tissue. In addition, the filum may adhere to other structures, which limits its normal compliance and may lead to signs of traction, even if the conus terminates at a normal level. Very thin sagittal T2-weighted (T2-W) or balanced steady-state free precession images may be necessary to image the filum.
3.2 Syringohydromyelia
Hydromyelia and syringomyelia are both characterized by the presence of fluid within the central cord. Hydromyelia involves the central canal, whereas syringomyelia does not. However, the pathogenesis of these abnormalities is similar, and they are clinically indistinguishable and frequently lumped together as “syrinx”, or syringohydromyelia. A congenital syrinx may develop from an obstructed fourth ventricle in the fetus [43], but a syrinx can also develop from posttraumatic/ischemic myelomalacia or obstruction due to adhesions, compression, or tumor. Syrinxes occur in 63 % of patients with myelomeningocele and scoliosis, and therefore, preoperative MRI is recommended prior to scoliosis instrumentation [44]. A syrinx often accompanies cord tethering, congenital hydrocephalus, and other congenital abnormalities. Hydrosyringomyelia is a common component of Chiari I malformations, probably due to obstruction. Occasionally, isolated primary hydromyelia becomes evident only in late childhood [45].
Imaging
If the syrinx is large, radiographs rarely may reveal fusiform enlargement of the spinal canal, with widening of the interpediculate distance and of the sagittal diameter of the canal. This may be limited to a portion of the spine (often cervical) but in severe cases extends inferiorly into the thoracic and even lumbar spine. MRI is necessary to evaluate the spinal cord, allowing differentiation of syrinx from, for example, a long-standing neoplasm. It can also demonstrate tonsillar herniation, indicating Chiari malformation.
3.3 Diastomatomyelia
This refers to a longitudinal split in the spinal cord that develops when the notochord divides during cell migration [11]. The two parts are usually asymmetric in size and are separated by a bony (Fig. 3.35), cartilaginous, or fibrous septum; the cord usually unites distally [46]. Diastematomyelia is most common in the upper lumbar region (Fig. 3.36) but can occur anywhere from the lower thoracic spine to the sacrum. Cervical and basicranial diastematomyelia are rare and usually associated with Klippel-Feil (see Fig. 3.40 below) and Chiari malformations [47].
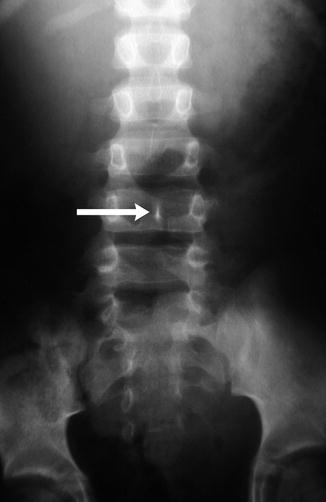
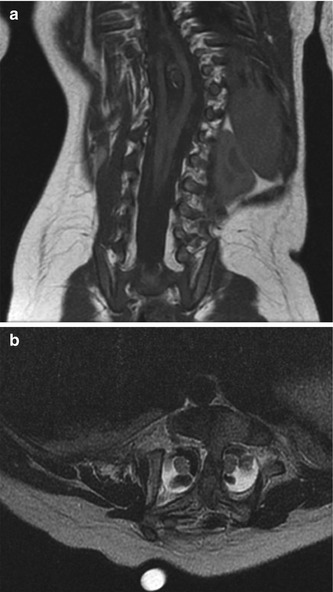
Fig. 3.35
Diastematomyelia with bony septum (arrow)
Fig. 3.36
Diastematomyelia. (a) Coronal T1-W image shows the two hemicords unite below a bony spur. (b) Axial fluid-sensitive sequence shows two hemicords
The conus is almost always low, and neurologic signs are likely related to tethering or stretching of the spinal cord. In up to 70 % of cases, each hemicord is encased in its own dural sheath. Skin changes are present in up to 80 % of patients and are similar to those found in other occult spinal dysraphisms. The location of the skin abnormality does not necessarily correlate with the septum level. Diastematomyelia should be distinguished from cord duplication or diplomyelia.
Because the notochord gives rise to vertebral bodies, diastematomyelia can be accompanied by neurenteric cysts, dorsal enteric fistulas, and abnormal vertebral bodies such as hemi- and/or butterfly vertebrae.
Imaging
Radiographs may demonstrate associated increased interpediculate distance and narrow intervertebral discs with possible vertebral body fusion. A subtle linear density may be apparent at the midline if the septum is osseous (see Fig. 3.35). CT depicts a bony septum but will identify the hemicords and a fibrous or cartilaginous septum only if CT myelography is performed. MRI depicts the cord anomaly as well as the dural sac(s) and demonstrates a fibrous, cartilaginous, or bony septum (see Fig. 3.36). The septum often extends anteriorly from the neural arch and thus may not be visibly fused to the vertebral body.
3.4 Congenital Intraspinal Tumors
Lipomas represent 1 % of all spinal cord tumors and may be intramedullary, extradural, or mixed; these lesions are often associated with meningocele, in which case they are termed lipomeningocele. Extravertebral lipomas may enter the spinal canal and cord or remain totally subcutaneous; associated anomalous bony structures may be present. Intraspinal lipomas are strongly associated with large sacral sacs, a low conus, and vertebral abnormalities. Occasionally, neural elements may enter these extraspinal tumors.
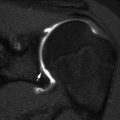
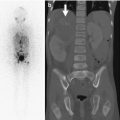
Sacrococcygeal teratomas (SCT) (Figs. 3.37 and 3.38) are the most common tumor of the fetus and newborn and are more common in girls [48]. They are closely associated with the coccyx and may involve the spinal canal. By definition they contain three germ cell layers (ectoderm, mesoderm, and endoderm). SCT may be mature or immature and may develop malignant transformation especially if internal (see staging below), in males, or if diagnosed after 2 months of age (Box 3.3). Fetal SCT may present with maternal increased fundal height and polyhydramnios, prompting imaging evaluation and diagnosis of the mass by US and subsequently MRI (Fig. 3.39) [48]. Prenatal hydrops due to a highly vascular tumor portends a poor outcome.
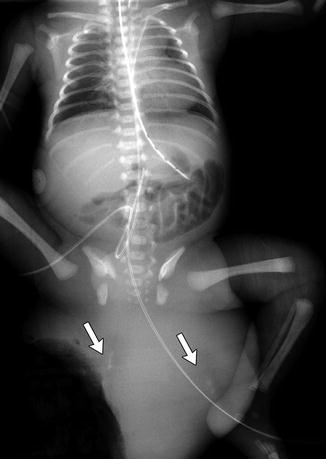
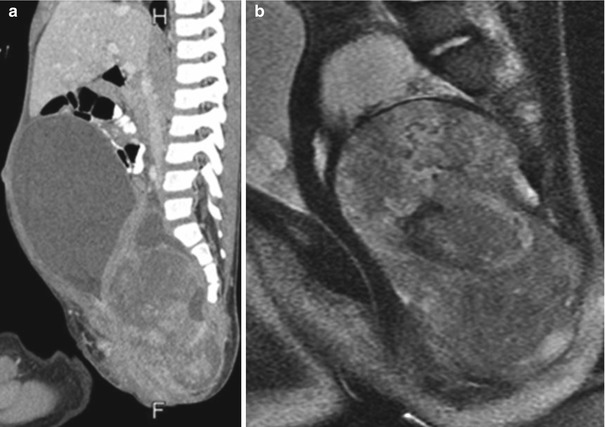
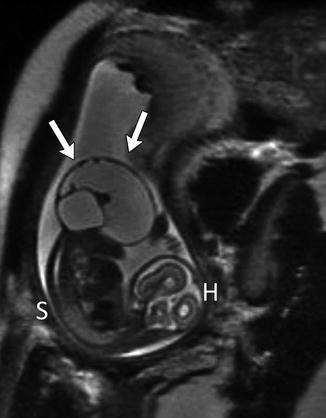
Fig. 3.37
Huge exophytic type I sacrococcygeal teratoma in a newborn boy. There are ill-defined calcifications within the mass (arrows) (Umbilical catheters are malpositioned)
Fig. 3.38
Type IV sacrococcygeal teratoma. Sagittally reformatted contrast-enhanced CT (a) and sagittal T2-W MRI (b) show a large, inhomogeneous, predominantly solid presacral mass, which elevates and obstructs the bladder
Fig. 3.39
Large exophytic predominantly cystic sacrococcygeal teratoma in a 30-week fetus (single-shot fast-spin echo MRI). Arrows delineate teratoma. H fetal head, S fetal spine
Newborns usually present with a mass. Associated genitourinary anomalies include hydronephrosis and renal dysplasia. Rectal atresia/stenosis or Currarino triad (presacral mass, scimitar sacrum, and anal malformation) may be present. Treatment is complete surgical resection, including resection of the coccyx.
Box 3.3: Sacrococcyngeal Teratoma (SCT)
Most common tumor of fetus/newborn |
Contains all three germ cell layers |
May be malignant, especially if detected after 2 m of age, in male, or completely internal |
Associated genitourinary abnormalities |
Heterogeneous mixed solid/cystic mass |
Sacrococcygeal teratomas may be classified [49] as type I, external (these account for half of the cases [50]) (see Fig. 3.37); type II, predominantly external with presacral internal extension; type III, external and internal with extension into the abdominal cavity; and type IV, entirely internal (see Fig. 3.38).
Imaging
Radiographs may show segmentation abnormalities in the case of lipomas and lipomyelomeningoceles or show a presacral or external SCT mass (see Fig. 3.37). Pre or postnatal US may identify presacral masses and characterize SCT as solid, cystic, or—most often—heterogeneous (15 % are purely cystic) [48]. The vascular/shunting nature of the solid portion of the tumor may be ascertained by Doppler interrogation. MRI provides greater anatomic detail for lipomas, lipomyelomeningoceles, and associated spinal cord abnormalities and may better delineate the extent and characteristics of internal SCT (see Fig. 3.38).
4 Syndromes with Spinal Dysplasia
4.1 Klippel-Feil Syndrome
In Klippel-Feil syndrome, the cervical and upper thoracic spine fuses partially or completely, forming a bony mass (Box 3.4). In its most extreme form, the neck is essentially absent [51]. However, the term can describe any degree of congenital cervical spine fusion if the clinical triad of short neck, low posterior hairline, and limited range of motion at the neck is evident. External and middle ear atresia and malformation may be present as well. Many patients have brain stem or high vertebral anomalies, making them vulnerable to cord impingement if manipulated.
Box 3.4: Klippel-Feil Syndrome
Clinical triad of short neck, low posterior hairline, and limited range of motion at neck |
Cervical and upper thoracic spine fusion and/or segmentation anomalies |
C2–3 and C5–6, the most commonly affected |
May also have rib fusion anomalies |
Sprengel anomaly in up to 50 % |
Renal anomalies in about 50 % |
Neurologic dysfunction is associated with increased mobility at unfused levels and may progress slowly or decompensate acutely after trauma. Spinal cord compression occurs either anteriorly in flexion or posteriorly due to an hypertrophied ligamentum flavum. Additionally, excess motion above or below fused levels may result in vertebral artery occlusion [52].
Twenty-five percent of patients with congenital scoliosis or kyphosis demonstrate structural malformations of the cervical spine, and 60 % of children with Klippel-Feil have scoliosis that exceeds 15° [53]. Undescended scapula, or Sprengel anomaly (see Chap. 5), occurs in up to 42 % of patients with Klippel-Feil syndrome. In up to half of children with Sprengel deformity, there is an omovertebral bone which extends from the medial border of the scapula to the posterior elements of C5–C7 [54].
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


