and Ashutosh Dash2
(1)
Nuclear Security and Isotope Division, Oak Ridge National Laboratory, OAK RIDGE, USA
(2)
Isotope Production and Applications Division, Bhabha Atomic Research Centre, Mumbai, India
17.1 Introduction
Radiopharmaceuticals are administered by parental routes and must comply with strictly controlled sterility requirements and, where relevant, aseptic working conditions are required for the manufacture of sterile medicinal products. The RPh manufacturing process must adhere to Quality Assurance (QA) requirements incorporating Good Manufacturing Practice (GMP) and thus Quality Control (QC) procedures which must not only be well documented, but which must also be effectively monitored (Cox et al. 1994). The cGMP compliance covers the manufacturing process and facility, quality guidelines, and personnel training. The term cGMP is used to define the latest best practice for the manufacture of pharmaceutical and allied products in various countries around the world (Decristoforo et al. 2008; Gouveia et al. 2015). Good manufacturing practice (GMPs) is that part of quality assurance which ensures that RPhs are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the product specification. The GMP is concerned with both production and quality control. In this chapter, the basic concepts of QA, GMP, and QC are interrelated and are described to emphasize their relationships and importance in the endeavor of radiopharmaceutical production and control.
17.2 The Radiopharmaceutical Manufacturing Process Elements
17.2.1 Quality Assurance (QA)
Quality assurance (QA) encompasses all parameters which individually or collectively concern the preparation and control of a finished product (Khurshid and Sadiq 1996). The QA criteria for radiopharmaceuticals include appearance, radiochemical purity/yield, radiochemical stability, radionuclide purity, radionuclide identity, assay for radioactivity, specific activity, pH, chemical purity, residual solvents, bacterial endotoxins (BET), and sterility (Cohen 1975). QA is of paramount importance for the manufacture of radiopharmaceuticals because of their unique characteristics, which include the administration of often very small sub-pharmaceutical mass, which in itself does not illicit a pharmaceutical response. In addition, RPhs are often administered in low volumes, and in some circumstances with ultra-short-lived radioisotopes (i.e., short-lived positron-emitting radioisotopes (11C, t 1/2 20 min; 18F, t 1/2 110 min, etc.), it is even necessary to administer the product before testing is complete. In these cases, extensive preclinical evaluation has substantiated the product sterility in multiple batches. QA is commonly broadly used for the confirmation and validity of various ways and measurements adopted to obtain a high-quality procedure with the objective of ensuring that pharmaceutical products are of the quality required for their intended use.
For these reasons, the manufacture of radiopharmaceuticals should ensure that they are designed and developed in a way that takes account of the requirements of GMP, good laboratory practice (GLP), and good clinical practice (GCP) (Decristoforo and Peñuelas 2009; Woldring 1999; De Vos et al. 2006; Otte and Maier-Lenz Dierckx 2005; Elsinga et al. 2010). The required production and control operations must be available in a clearly documented format and follow GMP requirements. The requirements for responsible individuals must be clearly specified and appropriate arrangements must be in place for the manufacture, supply, and use of the approved raw and packaging materials. All necessary controls must be conducted by well-defined and documented procedures for starting materials, including the radionuclide, intermediate products, and bulk products. These include in-process controls, calibrations, and validations. The radiopharmaceutical product must be correctly formulated and checked, according to defined procedures. Radiopharmaceuticals cannot be approved for commercial availability or supplied for patient use before an authorized individual has certified that each production batch has been produced and controlled in accordance with the requirements of the marketing authorization and any other regulations relevant to the production, control, and release of medicinal products. Satisfactory arrangements must also be developed to ensure, as far as possible, that RPhs are stored, distributed, and subsequently handled so that quality is sustained throughout their specified shelf life. In addition, procedures must be in place for self-inspection and/or quality audit to regularly appraise the effectiveness and applicability of the quality assurance system. Any deviations in any of these requirements are required to be reported, investigated, and recorded, and there are systems in place for approving changes that may have an impact on product quality. Finally, regular evaluation of the quality of radiopharmaceuticals should be conducted with the objective to authenticate the consistency of the process and ensuring its continuous improvement.
17.2.2 Good Manufacturing Practices (GMP) for Radiopharmaceuticals
The GMPs for radiopharmaceutical are the part of quality assurance that ensures that they are consistently produced and controlled in such a way to meet the quality standards appropriate for the intended use, as required by the approved specifications in the market authorization of the radiopharmaceutical in dosage form (Decristoforo et al. 2008; Woldring 1999; Elsinga et al. 2010; Gouveia et al. 2015). GMP basic requirements are as follows. With a view to ensure consistency as well as compliance with approved regulatory specifications, it is essential to clearly define and control the RPh production processes through which they are formulated and produced. In this case, the reproducibility of the production process is a principal criterion for ensuring consistency of quality, efficacy, and safety between different batches. Validation of critical formulation process steps must be carried out regularly as well as for any significant changes brought for processes improvement. The depth and scope of validation depend on the diversity, complexity, and criticality of the production process (Zigler 2009). Scientific and technical judgment should be used to justify the process changes and validation studies. All necessary key elements for GMP must be available, including the necessity of qualified and trained personnel, the availability of adequate premises and space, including appropriate hot cell facilities, remote handling equipment, and services. In addition, only approved radionuclides, excipients, and containers can be used for the formulation. Also, recommended procedures and instructions must be available and up-to-date and proper storage/quarantine facilities must be available.
Personnel involved with the production, quality control, and documentation of RPhs should have adequate training in accordance with applicable official federal, state, and local laws. Instructions and production procedures are required to be clear and written in unambiguous language. Written procedures should not only specify critical processing steps and factors (such as irradiated target processing time, temperature, and reagent purity, etc.) but must also specify the acceptance criteria, as well as the type of validation to be conducted (e.g., retrospective, prospective, or concurrent) and the product batch number/identification. These records are also required to address the identity of the batch and the amount of activity produced. The GMP facility must insure that all steps required by the recommended procedures and instructions are in place. It is also essential that qualified and competent operators are trained to document procedures. The key elements for GMP radiopharmaceutical production are depicted in Fig. 17.1.
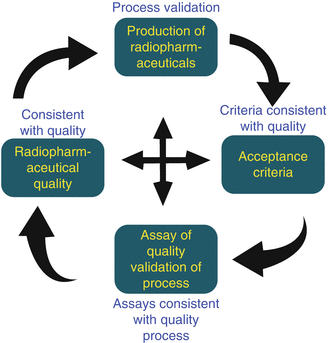
Fig. 17.1
Radiopharmaceutical GMP production
17.2.3 Quality Control (QC)
Quality control is that part of GMP which is concerned with sampling, specifications, and testing and with the organization, documentation, and release procedures which ensure that the necessary and relevant tests are actually carried out and that radiopharmaceuticals are not released for patient use until their quality has been assessed to be satisfactory (Cohen 1975; Cox et al 1994). QC is not confined to laboratory operations, but may be involved in many decisions to ensure the quality of the product. Basic requirements of quality control include that adequate facilities, trained personnel, and approved procedures are available to perform sampling, inspecting, and testing of starting materials, packaging materials, and intermediate, bulk, and finished products, and where appropriate for monitoring environmental conditions for GMP purposes. Samples of starting materials, packaging materials, intermediate products, bulk products, and finished products must be reserved by trained personnel and by methods approved by QC. Of course, all test methods require validation and appropriate records must be prepared and maintained, either electronically or by hard copy, which demonstrate that all the required sampling, inspecting, and testing procedures were actually carried out. Any deviations must be fully recorded and investigated. The final finished products containing the API (i.e., active pharmaceutical ingredient, Sect. 17.4) must comply with the qualitative and quantitative composition requirements for marketing authorization, including the desired purity, and enclosure within a suitably labeled appropriate container. As described earlier, a very important requirement is that records of inspection results are maintained and that testing of materials, intermediate, bulk, and finished radiopharmaceuticals is formally assessed against specification. Product assessment includes a review and evaluation of relevant production documentation and evaluation of deviations from specified procedures. Product batches cannot be released for use, sale, or supply prior to certification by an authorized individual, in accordance with the requirements of the relevant authorizations. Finally, sufficient reference samples of starting materials and products must be retained in the final package, unless exceptionally large packages are required for the product (i.e., large volume, high activity, heavy shielding, etc.), to permit future product examination.
17.3 Personnel
Radiopharmaceutical production is carried out in an interdisciplinary environment, including chemists with radiopharmacists, engineers, clinician-scientists, technologists, and biologists (Duatti and Bhonsle 2013; Lass and Scheffler 2003; Wang 1996). In light of the perceived need to establish and maintain a satisfactory quality assurance system, a staff of qualified and competent personnel is required to perform production, control, and supervision. These individuals must have the appropriate motivation, education, knowledge, and professional skills necessary to support the establishment and to perform the required tasks. Availability of well-trained skilled personal is critical to a viable RPh production program which must be conducted under the active direction and supervision of competent technical staff with prescribed qualifications and practical experience with competence in radiation protection. All responsible staff should have specific duties recorded in written descriptions and adequate authority to carry out their responsibilities without any gaps or unexplained overlaps. Personnel involved in the production of the radiopharmaceutical should receive training appropriate to the duties and responsibility assigned to them to ensure the safe manufacture of radiopharmaceuticals and radiation safety procedures, and regular in-service training is also required. Activities between radioactive and non-radioactive areas are permitted only if the safety rules of radiation control (health physics/radiation safety control) are respected, and only the minimum number of personnel required should be present in clean and aseptic areas when work is in progress. Access to these areas should be restricted during the preparation of radiopharmaceuticals, kits, or sterile set-ups. The number of personnel employed must also be adequate and in direct proportion to the workload and personnel involved in QA and QC operations must of course be suitably qualified and experienced. The duties of technical and quality control personnel must be defined, documented, and strictly adhered to. In order to avoid potential conflicts of interest, the individual responsible for the quality control laboratory must have responsibilities and activities which are independent of the manufacturing unit.
17.4 Active Pharmaceutical Ingredient (API)
Any substance or mixture of substances intended to be used in the manufacture of radiopharmaceuticals and used in the production of a radiopharmaceutical is defined as an API (Cervera-Padrell et al. 2012; Kumar et al. 1992). Such substances are intended to provide pharmacological activity or other direct effect in the diagnosis as well as treatment of disease or to affect the structure and function of the body. All radionuclides used in the manufacture of a radiopharmaceutical without a purification of the final preparation before administrations are considered an API. For radiopharmaceutical kits—which are targeting agents to which the radioisotope is attached—the active ingredient is considered to be that part of the formulation that is intended to carry or bind the radionuclide or to permit its binding.
Starting materials used in the radiolabeling reaction for the manufacture of RPhs which are purified after the radiolabeling process are not active pharmaceutical ingredients but are API starting materials. Because of this distinction, several rules need to be applied. For instance, all starting materials used in radiolabeling reactions for the preparation of radioactive pharmaceutical ingredients (APIs) that are not isolated and/or fully analyzed before incorporation in the final radiopharmaceutical preparation have to be analyzed according to a monograph of pharmacopoeia and must comply with the requirements of the pharmacopoeia. In case the substance is not described in a pharmacopoeia, it must be analyzed using validated methods. All analyses have to be performed in accordance with national regulations. If starting materials which are to be considered as APIs are produced by a foreign GMP-certified facility, a copy of the certificate of GMP production must be submitted to the competent authority of the user country for mutual recognition. Excipient starting materials used in the preparation of radiopharmaceuticals such as solvents, buffers, stabilizers, additives, antimicrobial agents, etc., must be of pharmaceutical l quality (as indicated on the label), be accompanied by a certificate of analysis, or be analyzed using validated methods and in accordance with national regulations.
The manufacture of all APIs should be carried out under general GMP requirement and although aseptic is not necessarily required to curtail the risk of contamination (i.e., US Food and Drug Administration). The emphasis on quality is most prominently manifested by the fact that all equipment, instruments, technologies, and accessories associated in the production of API must fulfill preset criteria and the product obtained has to meet strict specifications. Written and approved protocols specifying critical steps, acceptance criteria, must be in place. Confirmation of appropriate regulatory conditions for aseptic processing and its supportive activities are often mandatory.
17.5 Radionuclide Production
The procedure for the production of the radionuclide (see Chaps. 5, 6, 7, and 8) clearly describes major parameters, such as target material, nuclear reactions, construction and target holder material, and irradiation data—such as beam energy and intensity or neutron flux, etc. In addition, typical radionuclidic contaminants for the adopted procedure of separation/purification of the desired radionuclide and the effects on the efficiency of the production in terms of quality and quantity of the produced radionuclide must be evaluated. Radionuclide production for RPh formulation involves adherence to regulations on radiation protection. Therefore, the principles of qualification and validation should be applied to radionuclidic production, and a risk management approach should be used to determine the extent of qualification/validation, focusing on a combination of GMP and radiation protection.
17.6 Radiopharmaceutical Manufacture
The production of radiopharmaceuticals is a very specific and complex task required to meet quality standards to ensure product quality during all stages of the production process (Decristoforo et al. 2008). While undertaking the production of radiopharmaceuticals for human use, the availability of standard operating procedures (SOPs) must be considered at the RPh production site to ensure the regularly required reviews are kept up to date (Callahan et al. 2007). The purpose of an SOP is to carry out the operations correctly and always in the same manner. Specifications for starting materials used for the preparation of RPhs should include details of source, origin, methods of manufacture, and the controls used to ensure suitability for the intended use. Release of starting materials for the preparation of an RPh should be conditional and solely based on satisfactory results being obtained in the tests on starting materials. Adequate consideration must be given to the validation of sterilization methods. A list of critical equipment used for RPh production must be available, calibrated, or tested at regular intervals and should be checked before the initiation of production. In the case of radiolabeling kits, freeze drying must be carried out as an aseptic procedure, and the dispensing, packaging, and transportation of RPhs should comply with the relevant national regulations and international guidelines.
17.6.1 Sterile Production
Within the strictest definition of sterility, a specimen would be considered sterile only when the substance or material can be demonstrated to be completely absent of viable microorganisms (Brown and Baker 1996; Castronovo 1992; Chen et al. 1974). While a wide range of methods such as steam, dry heat, and filtration can be used for sterilization, it is important to note that the method used must both sterilize and maintain the strength, purity, and packaging integrity. Sterile RPhs may be divided into those which are manufactured aseptically and those which are sterilized at a terminal stage of the process. With a view to maintain sterility of an RPh which has been assembled from components, careful attention must be given to the environment, personnel, critical surfaces, container/closure sterilization and transfer procedures, the holding period of the RPh before dispensing into the vial, and any other key factors which may compromise sterility.
17.6.2 Terminal Sterilization
A RPh can also be sterilized in the final container after preparation and dispensing. This process is referred to as terminal sterilization and not only minimizes upstream bio-burden, but also reduces the challenge to the subsequent sterilization process. Terminal sterilization through membrane filtration is a process for the removal of any viable microorganism. In this process, the RPh formulation solution is filtered through one or two sterilizing-grade (0.22 μm) membrane filters into a sterilized receiving vessel located in a traditional clean room with an ISO Class 5 unidirectional airflow, isolator, or restricted access barrier system (RABS) to produce sterile effluent. For effective sterilization, the integrity of the sterilized filter must be verified and confirmed immediately after use by appropriate methods such as a Bubble Point, Diffusive Flow, or Pressure Hold Test (Belanger et al. 2009; Hayashi et al. 2014; Jornitz 2001).
17.6.3 Aseptic Sterilization
In this aseptic process, the RPh, containers, and closures are sterilized separately and then assembled under an extremely high-quality environmental condition designed to reduce the possibility of any nonsterile components. Aseptic sterilization presents a higher risk of microbial contamination of the product than terminal sterilization, and the principles of cGMP must be carefully observed during sterilization. In particular, the involvement of qualified personnel with appropriate training is required, along with adequate facilities, suitable production equipment—designed for easy cleaning and sterilization—adequate precautions to minimize bio-burden prior to sterilization, validated procedures for all critical production steps, environmental monitoring, and in-process testing procedures. Process validation includes appropriate regular checks on the process, by means of process simulation tests using microbial growth media that are then incubated and examined for microbial contamination (media fill tests). In addition, a suitable sample of each batch of any product that is sterilized by filtration and/or aseptically processed must be tested for sterility before the batch is released.
In this regard, several points need to be taken into account while undertaking RPh sterilization. First of all, the appropriate level of environmental cleanliness of the facility must be maintained. For the manufacture of sterile products where the working zone in which the products or containers may be exposed to the environment, cleanliness requirements should be complied. An essential aspect of contamination prevention is the adequate separation of areas of operation, and it is vital for rooms of higher air cleanliness to have a substantial positive pressure differential relative to adjacent rooms of lower air cleanliness. In addition to these points, a risk assessment analysis should be carried out to determine that appropriate pressure differences, air flow direction, and air quality are maintained in the clean room where radiopharmaceuticals are produced. The number of personnel in an aseptic processing room should be minimized, and both personnel and material flow should be regulated and minimized to prevent potential for the introduction of contaminants. The design of the equipment used in aseptic processing should limit the number and complexity of aseptic intervention by personnel, and the equipment should be appropriately designed to facilitate ease of sterilization. It is also important to ensure ease of installation to facilitate aseptic setup. Appropriate training should be conducted before an individual is permitted to enter the aseptic manufacturing area. Under the cGMP regulations, equipment must be qualified, calibrated, cleaned, and maintained to prevent contamination and mix-ups. The cGMP regulations place as much emphasis on process equipment as on testing equipment while most quality systems focus only on testing equipment.
Sterilization of RPhs by filtration must be conducted in a grade C environment, and if filtration sterilization is not performed, RPh preparation should then be carried out in a grade A environment with grade B in the background. Another important operational factor is minimization of the time between the initiation of RPh preparation and the filter sterilization or filtration stage through a microorganism retaining filter. There must also be a set maximum permissible time periods for each product that takes into account the composition and method of storage mentioned in the Master Formula record. In addition, all steps of the sterilization process should be appropriately validated and the validity of the process verified at regular predetermined intervals. Periodic bio-burden monitoring of products before terminal sterilization must also be performed and controlled to limits specified for the product in the Master Formula. Verification of the sterilized filter integrity is also a key point which must be confirmed immediately after use by appropriate methods such as Bubble Point, Diffusive Flow, or Pressure Hold Test. Record keeping at this stage is also important, and sterilization records must be maintained for each sterilization process. These processes may also include thermographs and sterilization monitoring strips, and all of these data must be maintained as part of the batch release procedure.
Any closures, containers, and stoppers should be designed to simplify cleaning and also to insure airtight seal of the RPh. The closures and stoppers should also be of such quality to avoid the risk of toxicity. As with any aseptic processing operation, it is critical that product contact surfaces should be sterile. A validated steam-in-place cycle, or equivalent process, should be used to sterilize the equipment path through which the product is conveyed. Prior to RPh production, assembly of sterilized equipment and consumable materials such as tubing, sterilized filters, and sterile closed and sealed vials to a sealed fluid path must be performed under aseptic conditions. While the manufacturing of sterile radiopharmaceuticals remains at the interface of various disciplines, sterility testing is a crucially important component and includes following microbiological laboratory controls, sampling and incubation, and investigation of sterility positives. The identification of organisms by sterility testing is important. The documentation of laboratory tests and deviations, monitoring of production area environment and personnel, product presterilization bio-burden, and production record review are also key components.
Microbiological controls and bacterial endotoxin testing (BET) must be performed according to written policies and procedures for specific compounded RPh (i.e., radiopharmacy preparation) when these tests are required as release criteria. Retrospective sterility testing—as in the case for use of ultra-short-lived radioisotopes—and BET should be conducted on randomly selected batches of the product to evaluate adequacy of the aseptic technique employed. These tests should also be conducted at regular periodic intervals, depending on historic results and trends, and should be completed more frequently when new personnel are involved. Sterility testing and BET should be performed according to locally accepted test methodology. The endotoxin limit for intrathecal RPh administration radiopharmaceuticals is significantly less than values allowed for intravenously administered RPhs (maximum administration of 14 EU vs. 175 EU, respectively). For this reason, a BET appropriate for detecting endotoxin concentrations at this lower limit should be performed by qualified personnel for each compounded radiopharmaceutical intended for intrathecal administration.
17.6.4 Sanitation and Hygiene
In the light of the perceived need to prevent possible contamination and cross contamination, a high level of sanitation and hygiene which is clearly outlined in SOPs must be practiced in every aspect of RPh production. Any residues and contaminants from previously manufactured products can cross-contaminate subsequent products from equipment or from the cleaning procedure (detergents/sanitizers) or degradation products resulting from the cleaning process itself. A high level of sanitation and hygiene insures product safety and purity and assures the quality of the process from an internal control and compliance points of view. The range of sanitation and hygiene includes personnel, premises, equipment and apparatus units, starting materials used for RPh production, products used for cleaning, de-sanitation and disinfection, and all other issues which could become a source of product infection or contamination. An integrated comprehensive sanitation and hygiene program must therefore be instituted to preclude potential sources of contamination.
Because of all these crucial issues and potential dangers, it is obligatory to insure that the RPh production facilities are free from any accumulated waste, dust, debris, and even other similar material RPhs. The facilities must be cleaned and maintained in an orderly manner which requires the availability of validated procedure which is maintained to assure the effectiveness and consistency of a cleaning process. RPh production areas should not be used as a general thoroughfare and not used for storage of materials, except for the material being processed. A routine sanitation program should cover both the specific areas to be cleaned and cleaning intervals. Cleaning procedures include equipment and materials to be used, and personnel assigned to and responsible for the cleaning operation must be properly recorded. The working and in-process storage spaces must be adequate to permit orderly and logical positioning of equipment and materials which necessary to reduce the risk of cross contamination. While undertaking RPh production, effective control of microbial contamination and particulate contamination are thus a critically important aspect of for production documentation. A cleaning validation program must thus be instituted in order to demonstrate that any residues of substance previously manufactured are consistently reduced to levels that are acceptable and that the cleaning procedure itself does not contribute unacceptable levels of residual materials to equipment. Written, approved, and routinely reviewed procedures are thus required which clearly define the assignment of responsibilities for sanitation. The cleaning schedules, methods, equipment, and materials to be used in cleaning and records should be available in sufficient detail and should be maintained. The cleaning documentation should include detailed description of the levels of cleaning as well as methods necessary to inspect and verify equipment and SOP. Where microbial contamination may be an issue, consideration should be given to the integrity of the vessel prior to manufacture.
17.7 Documentation
Documentation constitutes an indispensable component of a QA system related to all aspects of GMP and is intended to preclude the risk of error that purely verbal communication may introduce. The aim is to account traceability of each RPh preparation and provide an audit to trace an individual product for suspected defects. It ensures that all personnel concerned with manufacture are intimately aware of this information necessary to decide whether or not to release an RPh batch for patient use. The instructions and standard operating procedures (SOPs) of RPh preparation should be written and independently approved for each procedure or activity associated with the production. A specification should be available for each material used as well as for the final, dispensed radiopharmaceutical. All documents related to RPh manufacture should be prepared, reviewed, approved, and distributed according to written procedures. In order to achieve regulatory compliance, it is essential to have a full documentation system providing traceability of radiopharmaceuticals that include the following.
Related posts:
 Radionuclide Synovectomy: Treatment of Inflammation of the Synovial Joints
Radionuclide Synovectomy: Treatment of Inflammation of the Synovial Joints
 Introduction: Radiopharmaceuticals Play an Important Role in Both Diagnostic and Therapeutic Nuclear Medicine
Introduction: Radiopharmaceuticals Play an Important Role in Both Diagnostic and Therapeutic Nuclear Medicine
 Inhibition of Arterial Restenosis Following Balloon Angioplasty
Inhibition of Arterial Restenosis Following Balloon Angioplasty
 Radioimmunotherapy (RIT)
Radioimmunotherapy (RIT)
 Therapeutic Radiopharmaceuticals for Bone Pain Palliation
Therapeutic Radiopharmaceuticals for Bone Pain Palliation
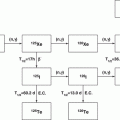 Reactor-Produced Therapeutic Radionuclides
Reactor-Produced Therapeutic Radionuclides
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


