- •
One great vessel and one semilunar valve arising from the heart.
- •
Origin of the branch pulmonary arteries from the trunk—from a single main pulmonary artery? Independently from the sides of the trunk?
- •
Commonly, no ductus arteriosus is present, unless the branch pulmonary arteries are discontinuous, in which case, the branch pulmonary artery not connected to the trunk is perfused by a ductus arteriosus.
- •
Large ventricular septal defect beneath the large single great vessel arising from the heart.
- •
Function of the truncal valve—is there stenosis or insufficiency?
- •
Aortic arch sidedness.
- •
Continuity or interruption of the aortic arch.
Anatomy and Anatomical Associations
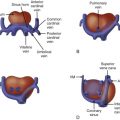
Truncus arteriosus (TA) is a conotruncal malformation of the heart. The embryologic conotruncus consists of the muscularized conus or conal septum and the adjacent truncus arteriosus, which gives rise to the outflow tracts. Early in development, the conotruncus originates from the primitive right ventricle but then shifts leftward over the forming ventricular septum. To complete normal development, septation of the truncus arteriosus must occur followed by rotation of the outflow tracts giving rise to a left ventricle–to–aorta relationship and a right ventricle–to–pulmonary artery relationship. In TA, septation does not occur properly, leaving a single arterial trunk that originates from both ventricles and gives rise to the aortic arch, at least one pulmonary artery, and at least one coronary artery. The arterial trunk almost always sits over a large ventricular septal defect (VSD), which exists because of a deficiency of the conal septum ( Figure 17-1 ).

TA occurs in several forms described in two main classification schemes. Collett and Edwards proposed a classification system in 1949 based on the origin of the pulmonary arteries. A type I TA occurs when a short main pulmonary artery trunk originates from the proximal trunk and gives rise to the branch pulmonary arteries. In type II TA, the branch pulmonary arteries arise separately from the arterial trunk but are close in proximity. In type III TA, the branch pulmonary arteries arise separately but are further away from one another. Type IV TA was originally described as pulmonary arteries originating from the descending aorta, but this anatomical variant is now considered to be a form of tetralogy of Fallot with pulmonary atresia. Van Praagh and Van Praagh proposed a modified scheme in 1965 ( Figure 17-2 ). A type AI is identical to the Collett and Edwards type I with a main pulmonary artery arising from the arterial trunk. Type AII includes all cases in which the branch pulmonary arteries arise separately from the proximal trunk regardless of the distance between them. Type AIII includes cases in which one pulmonary artery, usually the right, arises from the proximal trunk, and the contralateral lung is supplied by a pulmonary artery arising more distally either from the aortic arch or from a collateral vessel. Type AIV is TA associated with an interrupted aortic arch. In this type, a large common trunk gives rise to (1) a main pulmonary artery that bifurcates into both branch pulmonary arteries, (2) an ascending aorta that gives rise to head and neck vessels, and (3) a patent ductus arteriosus that connects to the descending aorta. Types AI and AII are the most common variants, followed by type AIV then AIII.

There is a single semilunar truncal valve in TA. This common truncal valve is often abnormal with thickened, dysplastic leaflets and is in fibrous continuity with the mitral valve. The truncal valve may have multiple leaflets, reflecting its developmental origins and potential for differentiation into the aortic and pulmonic valves, which failed to occur. A trileaflet truncal valve is most common, followed by a quadrileaflet and a bileaflet, or bicuspid, variant. Significant stenosis or regurgitation is common and can affect clinical presentation and prognosis. A ductus arteriosus is absent in Van Praagh type AI and AII, but is always present in type AIV, and can be present in type AIII because a pulmonary artery can originate from the ductus arteriosus. Coronary artery abnormalities are frequent, with a single coronary artery the most common variant. Other cardiac associations include aberrant origin of the arch vessels, right aortic arch, and a left superior vena cava. Intracardiac abnormalities such as hypoplasia of an atrioventricular valve and ventricular hypoplasia have been reported in association with TA, but these are exceedingly rare.
Frequency, Genetics, and Development
TA is an uncommon anomaly, accounting for 1% to 2% of all forms of congenital heart disease. Extracardiac anatomic anomalies and chromosomal abnormalities exist in nearly one half of patients with TA. The most common genetic abnormality is microdeletion of chromosome 22q11 (22q11 del). This is the chromosome abnormality identified in 90% of patients with DiGeorge syndrome and is associated with multiple extracardiac defects such as palatal abnormalities; feeding and speech difficulties; thymic hypoplasia leading to immunodeficiency and hypocalcemia; varying degrees of developmental, cognitive, and psychological deficits; facial dysmorphism; and renal and skeletal abnormalities. Congenital heart disease is estimated to occur in 75% to 80% of patients with 22q11 del; the most common cardiac defects noted are abnormalities of conotruncal formation, including tetralogy of Fallot, interrupted aortic arch, and truncus arteriosus. Over one third of patients with TA have been identified to have 22q11 del, and 50% of cases with an associated interrupted aortic arch (Van Praagh type AIV) have 22q11 del. The molecular and genetic causes specific to the formation of TA are still mostly unknown, although studies have demonstrated that abnormalities of neural crest cell migration play an important role.
Prenatal Physiology
In TA, the normal fetal flow patterns are disrupted. In the normal fetal heart, the left ventricle and aorta have higher oxygen content due to right-to-left streaming of ductus venosus flow across the atrial septum. The presence of a large VSD allows intracardiac mixing of the blood returning to the right heart, and with only one outlet in existence, the blood in the pulmonary circulation has the same oxygen content as the blood in the systemic circulation. In general, the fetus tolerates this environment quite well. However, significant abnormalities of the truncal valve can result in fetal distress in TA because the common arterial trunk is the only exit from the heart. Stenosis and regurgitation of the truncal valve tend to occur mutually within the same patient, typically, in the setting of a markedly dysplastic valve. In such cases, the fetus can develop ventricular dilation, heart failure, and hydrops.
If the pulmonary arteries are discontinuous as in TA Van Praagh type AIII, the pulmonary vasculature of the lung that is not attached to the main trunk may have relatively limited blood flow in comparison with the pulmonary vasculature of the lung that is directly receiving blood flow from the trunk. Such disparity in flow between the lungs may cause differences in pulmonary vascular development, which may be problematic after birth.
Intrauterine growth retardation is commonly present in the fetus with TA and is associated with 22q11del.
Prenatal Management
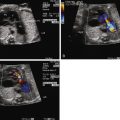
Fetal echocardiographic imaging will detail all elements of the anatomy. The four-chamber view may appear normal because the right and left ventricles and atrioventricular valves are of normal size and structure. A single great vessel with a single semilunar valve will be seen originating from the heart, with a large VSD beneath. The greatest challenge in TA is identifying the origin and course of the branch pulmonary arteries. The pulmonary artery origins should be identified in every patient in order to appropriately classify the type of TA and determine whether postnatal prostaglandin therapy should be initiated. The base of the trunk should be identified in either long- or short-axis view and a slow, careful sweep cephalad should reveal the origin of the pulmonary arteries. Alternatively, if not found in this manner, a reverse search back from “lung-to-trunk” may be helpful. By identifying the branch pulmonary artery in the lung parenchyma using color Doppler imaging, one can track the branch pulmonary artery vessel back to the heart and trace its origin from the trunk.
The differential diagnosis of TA includes tetralogy of Fallot with pulmonary atresia. In the latter, a separate main pulmonary artery trunk can be identified, even if there is no flow into it. In TA, at least one of the branch pulmonary arteries is seen arising from the trunk, whereas in tetralogy of Fallot with pulmonary atresia, no pulmonary artery arises directly from the aorta without an interposed ductus arteriosus or collateral vessel (see Chapter 11 ).
In the absence of severe truncal valve abnormalities or extracardiac abnormalities, the fetus with TA is usually stable. Serial fetal echocardiography studies are recommended to delineate the morphology and competency of the truncal valve and, if abnormalities are present, to monitor for signs of heart failure or hydrops. Color Doppler flow across the valve as well as pulse Doppler will allow an estimate of the degree of regurgitation or stenosis present.
Given the high frequency of extracardiac abnormalities in the TA population, a comprehensive obstetrical fetal anatomy ultrasound should be performed in every fetus, and an assessment of karyotype is strongly recommended. Counseling of families regarding the operative course and prognosis will be linked to these findings. Families should also be aware that there is the risk of fetal demise when there are truncal valve abnormalities.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


