Abstract
Turner syndrome (monosomy X) is characterized by the complete or partial loss of the second X chromosome in the female, with or without cell line mosaicism. There are three phenotypes: first-trimester abortus, second-trimester hydrops fetalis, and, least commonly, a neonate with a cystic hygroma. Turner syndrome is the most common sex chromosome abnormality and is the only monosomy that is compatible with life and has wide phenotypic variability. The incidence of Turner syndrome is 1 : 2000 to 1 : 2500 live-born females and it is not associated with an increased recurrence risk. Common prenatal ultrasound findings are increased nuchal translucency (NT), cystic hygroma, hydrops fetalis, cardiac or renal anomalies, and fetal growth restriction. Phenotype varies, and is related to the underlying karyotypic abnormality. Complete monosomy X (45,X) is more severe than cell line mosaicism. Spontaneous abortion occurs in greater than 90% of conceptuses with Turner syndrome. Patients with Turner syndrome are at risk of endocrine, autoimmune, cardiovascular, renal, and multiple other complications, and care should be coordinated with appropriate experts.
Keywords
Turner syndrome, cystic hygroma, hydrops, monosomy, sex chromosome abnormality
Introduction
Turner syndrome (monosomy X) is characterized by complete or partial loss of the second X chromosome in the female, with or without cell line mosaicism. There are three distinct phenotypes: (1) first trimester abortus (98%), (2) second trimester hydrops fetalis (often resulting in fetal demise), and least commonly (3) neonate with a cystic hygroma (thin-walled cyst containing lymphatic fluid). Turner syndrome is the only monosomy that is compatible with life and has wide phenotypic variability.
Disorder
Turner Syndrome
Definition
The phenotypic pattern of Turner syndrome was described in 1938 and defined cytogenetically in 1959 as an absent or abnormal second sex chromosome. Individuals with Turner syndrome always have short stature, typically have a webbed neck, and often have lymphedema, cardiac malformations, renal malformations, gonadal dysgenesis, and/or endocrinopathies. Many of these findings can be identified prenatally with ultrasound (US).
Prevalence and Epidemiology
Turner syndrome is the most common sex chromosome abnormality, and the second most common chromosomal abnormality associated with spontaneous abortion. Its true prevalence is difficult to ascertain because mild phenotypes may go undiagnosed. Best estimates are that it occurs in 4–5 : 10,000 live-born females. Turner syndrome is not associated with advanced maternal or paternal age, and has no known ethnic or racial influences.
More than 90% of conceptions with Turner syndrome spontaneously abort. Karyotypic analysis of products of conception from spontaneous pregnancy losses demonstrates an approximate 10% prevalence of 45,X. Prenatal prevalence, estimated through evaluation of cytogenetic registries, is about 40 : 10,000 in the late first trimester by chorionic villus sampling, and 17.5 : 10,000 in the second trimester by amniocentesis.
Etiology and Pathophysiology
Distinct etiologies are responsible for the various Turner syndrome karyotypes. Complete monosomy (45,X), the most common etiology of Turner syndrome, is due to absence of one sex chromosome, usually the paternal (80%).
More than 50% of individuals with Turner syndrome have a mosaic chromosomal complement, such as 45,X/46,XX. In the case of cell line mosaicism, the loss of genetic material occurs after fertilization. Mosaicism can also result from a cell division error, resulting in one cell line that exhibits a 45,X karyotype and a second line containing a 46,XX, 46,XY, or 47,XXX karyotype. Less commonly, there may be a structurally altered second X chromosome, or some cells carry a Y chromosome, resulting in a virilized adolescent. Individuals who are mosaics for sex chromosomes and/or have structurally abnormal karyotypes appear female and usually have fewer anomalies than the 45,X phenotype.
Postnatal karyotype of 200 patients with phenotypic Turner syndrome demonstrated 46% were 45,X, 41% had a second structurally abnormal X chromosome, 7% were 45,X/46,XX or 45,X/46,XX/47,XXX mosaics, and 6% had a structurally abnormal Y chromosome. In contrast, in 90% of prenatally diagnosed Turner syndrome, the karyotype is 45,X ( Table 152.1 ).
| Karyotype | Number of Cases | % |
|---|---|---|
| 45,X | 76 | 90.4 |
| 45,X/46,XX | 5 | 5.9 |
| 45,X/47,XXX | 1 | 1.2 |
| 45,X/46,XY | 1 | 1.2 |
| 45,X/46,XX/47,XXX | 1 | 1.2 |
| Total | 84 | 100 |
Clinical manifestations appear to result from a reduced complement of genes normally expressed on the randomly inactivated X chromosome and therefore karyotype alone cannot predict phenotype. Short stature is the only clinical finding invariably associated with 45,X individuals although, while this has been associated with a pseudoautosomal deletion encompassing a homeobox gene (short stature homeobox-containing gene on the X chromosome, SHOX ), this deletion is also present in 2% to 15% of cases of idiopathic short stature.
Common clinical manifestations, such as webbing of the neck, shield chest, renal anomalies, and cardiac defects may be a consequence of lymphedema interfering with organ development. For instance, it has been proposed that lymphatic obstruction at the level of the ascending aorta may directly cause flow-related cardiac defects such as bicuspid aortic valve and/or aortic coarctation as well as neck webbing.
Manifestations of Disease
Clinical Presentation
Characteristics of the 45,X phenotype are listed in Table 152.2 . The most consistent feature is short stature. Ovarian dysgenesis and renal anomalies, such as horseshoe kidney, ureteral duplication, and unilateral renal agenesis, are also relatively common. Cardiovascular abnormalities, which are seen more in 45,X individuals than in mosaics, were present in 44% of 173 patients with Turner syndrome in one series. Of these, 34% involved the aortic valve, 19% the aortic arch, 8% the systemic venous system, 5% the ventricular septum, and 4% the pulmonary venous system. Mental retardation is not normally seen unless a small ring X chromosome is present.
| Feature | Incidence (%) |
|---|---|
| Small stature | 100 |
| Ovarian dysgenesis | 90+ |
| Transient congenital lymphedema | 80+ |
| Shield chest and widely spaced nipples | 80+ |
| Prominent auricles | 80+ |
| Low posterior hairline | 80+ |
| Webbing of posterior neck | 50 |
| Anomalies of the elbow, including cubitus valgus | 70 |
| Hypoplastic or hyperconvex nails | 70 |
| Renal anomalies | 60+ |
| Short metacarpal and/or metatarsal | 50 |
| Hearing loss | 50 |
| Cardiac anomalies | 20+ |
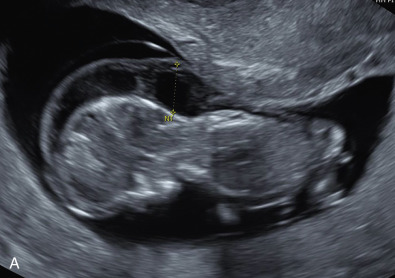
Two-thirds of cases are suspected from US findings. Classic US findings in the first trimester include increased NT, cystic hygroma, and hydrops fetalis ( Figs. 152.1, 152.3, 152.5 ). Although these findings increase risk for many aneuploidies, persistence of cystic hygroma is highly correlated with Turner syndrome.

Imaging Technique and Findings
Serum Analytes.
Serum analytes are not consistently abnormal, nor is there an indicative pathognomonic pattern when they are abnormal. However, the presence of a cystic hygroma or hydrops increases the sensitivity of serum analytes; second-trimester multiple marker screening is abnormal in 60% of cases with a cystic hygroma versus 11% of Turner cases without a cystic hygroma. Furthermore, fetuses with, but not those without, hydrops demonstrate elevated levels of inhibin A and beta-human chorionic gonadotropin.
Ultrasound.
The Turner phenotype refers to neonates, but many findings can be suspected and/or detected prenatally with US. Congenital anomalies are detected in about two-thirds of prenatally diagnosed Turner syndrome, and the classic finding is cystic hygroma ( Figs. 152.1 and 152.2 ; Table 152.3 ). Central lymphatic obstruction in the form of a thickened NT, cystic hygroma, or subcutaneous edema is often apparent (see Figs. 152.1 and 152.2 ). Hydrops fetalis also commonly develops secondary to an absent connection between the lymphatic and venous system, resulting in effusion in potential spaces (thoracic, pericardial, and abdominal) ( Figs. 152.3 and 152.4 ). The hygroma may resolve if a connection is later established restoring lymphatic flow, but this leaves a webbed neck and prominent nuchal skin folds.