Summary of Key Points
- •
Central nervous system (CNS) anomalies are among the most frequent malformations encountered by antenatal sonography.
- •
Ultrasound allows detection of a large proportion of all CNS malformations in early gestation. However, in some cases the findings may be subtle, whereas in others the anomaly may be progressive and manifest only in late gestation or after birth. Ultrasound will also frequently demonstrate normal anatomic variations difficult to differentiate from true malformations.
- •
Magnetic resonance imaging (MRI) is often useful with CNS anomalies to improve diagnostic accuracy.
- •
One of the most common antenatal diagnoses, enlargement of the lateral cerebral ventricles or ventriculomegaly, may be associated with other CNS malformations and requires a detailed workup.
- •
Neural tube defects, such as anencephaly and open spina bifida, are often recognized in early gestation. Demonstration of the spinal defect in spina bifida may be difficult, but the diagnosis is facilitated by frequently associated abnormalities of intracranial anatomy.
- •
Alobar and semilobar holoprosencephaly may be recognized in early gestation; more subtle varieties, such as the lobar type, are more difficult to identify.
- •
Complete agenesis of the corpus callosum (ACC) can be recognized by midgestation. Counseling regarding prognosis is difficult because the prognosis is variable and studies suggest that many infants diagnosed in utero may have normal intelligence. Partial agenesis is much more difficult to identify by prenatal sonography.
- •
Cystic and cyst-like anomalies of the posterior fossa are often difficult to diagnose with certainty. Although the increased fluid is easily recognized, a specific diagnosis is often difficult because many different entities exist and there is much overlap with normal variants.
CNS malformations are among the most common congenital abnormalities encountered at birth. The true incidence of these anomalies, however, is probably underestimated; most epidemiologic surveys are based upon clinical examinations performed in the neonatal period, and many cerebral malformations will only be discovered later in life. Long-term follow-up studies suggest that the true incidence may be as high as 1 in 100.
Detection of CNS anomalies was one of the early goals of sonography in pregnancy. Although there has been much debate regarding the value of ultrasound screening, it is now clear that open neural tube defects, including anencephaly, open spina bifida, and large cephaloceles, as well as other severe malformations such as holoprosencephaly, are easily identified in early gestation. Improvements in image quality have led to increased detection of relatively minor malformations, such that the challenge sonologists now face is not sensitivity, but rather specificity. Many CNS anomalies that are poorly understood (ACC and posterior fossa cysts are representative examples) are now detected antenatally and for many such abnormalities, the postnatal consequences are difficult to predict. Furthermore, there is often significant overlap between normal variation and pathologic changes, as with enlargement of the ventricular system and small dimensions of the fetal head. Fetal sonography can precisely diagnose some lethal or severe malformations. But in a significant proportion of patients (e.g., enlargement of the lateral ventricles alone has an in utero prevalence of 1%), CNS findings carry uncertain prognostic significance that can provoke much anxiety in expectant couples, require difficult decisions, and may eventually result in the loss of normal fetuses. It has been suggested that MRI may add important diagnostic and prognostic information, although the precise impact of this technique continues to be debated.
Embryology
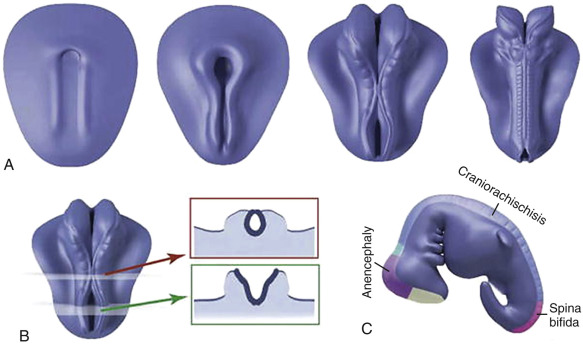
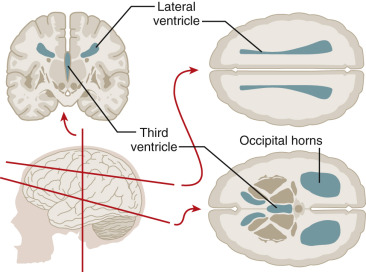
The nervous system develops from the neural plate, a thickened area of embryonic ectoderm. It is the notochord and paraxial mesoderm that induce the overlying ectoderm to differentiate into the neural plate. The neural plate folds along its central axis to form a neural groove lined on each side by a neural fold. The two neural folds fuse together and pinch off to become the neural tube. Fusion of the neural folds begins in the middle of the embryo and moves cranially and caudally. The cranial open end of the tube is the anterior (rostral) neuropore, and the caudal open end of the tube is the posterior (caudal) neuropore ( Figs. 9-1 and 9-2 ). The rostral neuropore closes on or before day 26 and the caudal neuropore closes 2 days later. The walls of the neural tube thicken to form the brain and spinal cord. The neural canal of the neural tube is converted into the ventricular system of the brain and the central canal of the spinal cord.
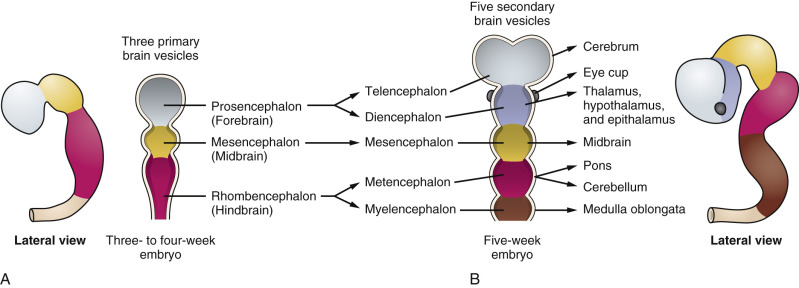
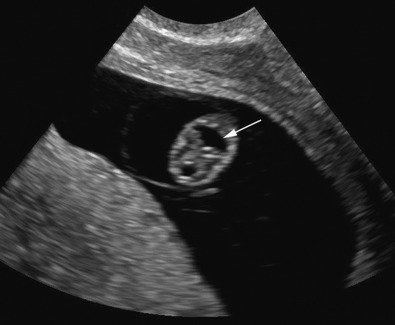
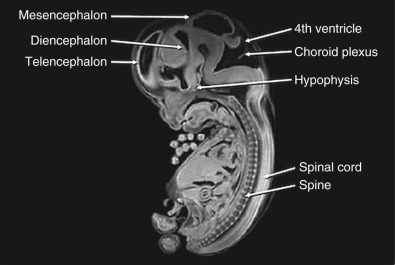
At about 8 to 10 weeks’ gestation, a focal dilatation of the neural tube is seen in the dorsal aspect of the developing hindbrain; it is the rhombencephalic vesicle, the predecessor to the fourth ventricle. This structure may appear quite prominent and should not be misinterpreted as representing ventriculomegaly or another CNS anomaly ( Figs. 9-3 and 9-4 ).
Ventriculomegaly
Enlargement of the lateral cerebral ventricles, commonly referred to as ventriculomegaly ( Fig. 9-5 ), is a nonspecific marker of abnormal brain development. The presence of normal fetal lateral ventricles on ultrasound examination decreases the risk of a CNS anomaly, whereas detection of ventriculomegaly increases the risk that a significant malformation is present. Although many different approaches to the evaluation of the lateral ventricles have been proposed, measurement of the width of the atrium (or posterior horn) of the lateral ventricle is currently favored. In a normal fetus, the measurement of the lateral ventricle should be less than 10 mm between 15 and 40 weeks’ gestation. In most fetuses with significant ventricular enlargement, the ventricles are symmetrically affected whereas cases with milder degrees of ventriculomegaly are more commonly unilateral. Male fetuses have a slightly larger atrial size than female fetuses. A study by Patel and associates demonstrated that atrial size demonstrated a near-normal distribution, with mean size for all subjects 6.1 mm ± 1.3 (standard deviation [SD]). When separated by sex, the mean atrial diameter of 122 female fetuses was 5.8 mm ± 1.3, and the mean atrial diameter of 97 male fetuses was 6.4 mm ± 1.3. The difference in mean size was statistically significant ( P < 0.005). Thus, a ventricular atrial measurement of 10 to 11 mm will have more significance in a female fetus than in a male fetus.
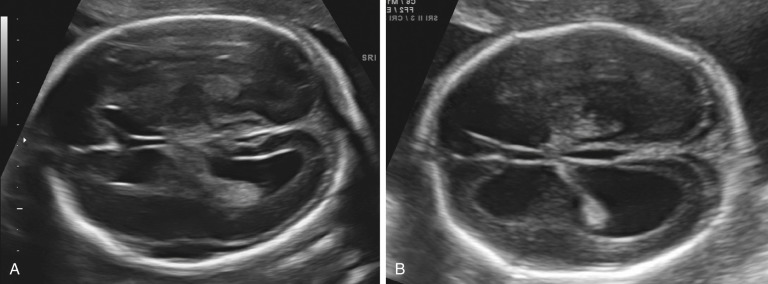
A ventricular atrial width of more than 15 mm indicates severe ventriculomegaly . This is almost always associated with an intracranial malformation, although the outcome is variable and depends largely upon the underlying cause. The available studies suggest that fetuses with isolated severe ventriculomegaly have an increased risk of perinatal death and a probability of severe long-term neurologic sequelae in at least 50% of survivors.
An intermediate value of the atrial width, 10 to 15 mm, is commonly referred to as mild ventriculomegaly and is less frequently associated with significant CNS anomalies, although it can be seen with ACC and open neural tube defects. Mild ventriculomegaly is also associated with chromosomal aberrations: the risk of trisomy 21 is increased 3.8 times when ventricular enlargement is an isolated finding. Fetal infections, such as cytomegalovirus (CMV), may result in ventricular enlargement, although usually other sonographic abnormalities are also present (areas of focal cerebral increased echogenicity, microcephaly, and porencephaly). When mild ventriculomegaly is isolated, and associated anomalies are absent, most infants are asymptomatic after birth. However, several reports have indicated that some fetuses are found to have severe CNS anomalies in advanced gestation or after birth (hydrocephalus, white matter injury, and cortical plate abnormalities), and some studies have reported an increased risk of neurologic compromise. Studies reporting on the prognosis of mild ventriculomegaly have been limited by the lack of standardized follow-up protocols and difficulty in defining “normal” neurologic outcome. However, most fetuses with mild ventriculomegaly will have a normal outcome, whereas about 10% demonstrate neurodevelopmental abnormalities of variable types and magnitude.
When mild ventriculomegaly is encountered, it is important to rule out associated anomalies. A targeted sonographic survey of fetal anatomy should be performed, with particular attention to details of the CNS. Fetal diagnostic testing for aneuploidy should be offered, and chromosomal microarray should be considered. Detailed ultrasound assessment of the CNS may be enhanced by transvaginal scanning, particularly if the fetus is in cephalic presentation. There is some debate as to whether fetal MRI is indicated in all cases of apparently isolated ventriculomegaly. This technique may provide significant diagnostic information, as it allows more detailed assessment of the developing fetal brain and detection of disorders of sulcation, cortical malformations, and migrational abnormalities. In continuing pregnancies in which mild ventriculomegaly has been identified, standard obstetric management is recommended.
Neural Tube Defects
The incidence, cause, and recurrence risk of neural tube defects are reported in Tables 9-1 through 9-3 . There are several different forms of neural tube defects.
| Geographic Area | Spina Bifida Incidence per 1000 Births | Anencephaly Incidence per 1000 Births |
|---|---|---|
| South Wales | 4.1 | 3.5 |
| Southampton | 3.2 | 1.9 |
| Birmingham, UK | 2.8 | 2.0 |
| Charleston | ||
| White | 1.5 | 1.2 |
| Black | 0.6 | 0.2 |
| Alexandria | 0 | 3.6 |
| Japan | 0.3 | 0.6 |
| Multifactorial Inheritance |
| Anencephaly, myelomeningocele, meningocele, and encephalocele |
| Mendelian Syndromes |
|
| Chromosome Abnormalities |
|
| Probably Hereditary but Mode of Transmission Not Established |
|
| Teratogens |
|
| Maternal Predisposing Factors |
|
| Specific Phenotypes, Without Known Cause |
|
| Population | Incidence per 1000 Live Births |
|---|---|
| Mother as Reference | |
| General incidence | 1.4-1.6 |
| Women undergoing amniocentesis for advanced maternal age | 1.3-3.0 |
| Women with diabetes mellitus | 20 |
| Women on valproic acid in first trimester | 10-20 |
| Fetus as Reference | |
| One sibling with NTD | 15-30 |
| Two siblings with NTD * | 57 |
| Parent with NTD | 11 |
| Half sibling with NTD | 8 |
| First cousin (mother’s sister’s child) | 10 |
| Other first cousins | 3 |
| Sibling with severe scoliosis secondary to multiple vertebral defects | 15-30 |
| Sibling with occult spinal dysraphism | 15-30 |
| Sibling with sacrococcygeal teratoma or hemartoma | ~15-30 |
* Risk is higher in British studies. Risk increases further for three or more siblings or combinations of other close relatives.
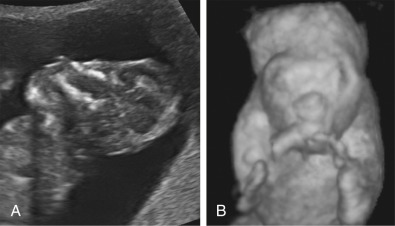
Anencephaly is characterized by the absence of the cranial vault and telencephalon. The diagnosis is made by ultrasound in the second and third trimesters and relies upon demonstration of the absence of the cranial vault. In addition, most cases can be confidently identified by 11 to 13 weeks’ gestation. At this time, the fetal head can be recognized as overtly abnormal owing to lack of an ossified calvarium ( Fig. 9-6 ). The terms acrania and exencephaly have also been used to describe this appearance; these represent early stages in the development of anencephaly. The outcome of anencephaly is uniformly fatal.
Spina bifida can occur in open as well as closed forms. Open spina bifida is characterized by a full-thickness defect of the skin, underlying soft tissues, and vertebral arches, with exposure of the neural canal. These defects may vary considerably in size, with the lumbar and sacral areas most frequently affected. Leakage of cerebrospinal fluid through the defect results in an increased concentration of alpha-fetoprotein (AFP) and acetylcholinesterase in the amniotic fluid and maternal serum, and measurement of maternal serum and amniotic fluid AFP can be used to screen for neural tube defects.
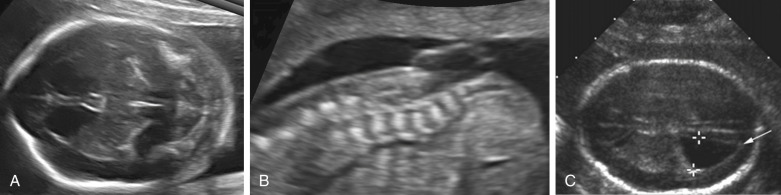
Open spina bifida can be identified sonographically by demonstrating the opening of the neural tube and the defect in the vertebrae (splaying outward of the posterior elements) as well as the overlying soft tissues ( Fig. 9-7 ). A cyst formed by the fusion of the malformed cord and meninges (myelomeningocele) is usually found. In a minority of cases, there is no covering membrane (myelocele). The diagnosis of a neural tube defect may be difficult and requires meticulous scanning. Examination of the fetal head is of great utility, as open spina bifida is consistently associated with characteristic and readily recognizable intracranial lesions. Leakage of cerebrospinal fluid leads to displacement of the cerebellum and medulla oblongata through the foramen magnum inside the upper cervical canal (Chiari type II or Arnold-Chiari malformation). Sonographically, this results in small head measurements in the midtrimester, as well as obliteration of the cisterna magna; small size and abnormal shape of the cerebellum (banana sign), which is located deep in the posterior fossa; scalloping of the frontal bones (lemon sign); and ventriculomegaly. Another characteristic abnormality is a “pointed” appearance of the occipital horns of the lateral ventricle. The occipital point is a common supratentorial feature of the Chiari II malformation. It is seen more commonly in fetuses younger than 24 weeks and in those with normal-sized ventricles. Interestingly, it is seen as commonly among fetuses with mild posterior fossa deformations as in those with more severe distortions (see Fig. 9-7 ). Overall, the detection rate of open spina bifida at the midtrimester obstetric sonogram has been reported to be at least 90%. Abnormalities of the posterior fossa and lateral ventricles have also been described at 11 to 13 weeks’ gestation, but the diagnostic accuracy of these findings to screen for spina bifida is still being assessed.
Several other neural axis abnormalities are frequently associated with myelomeningocele and Chiari II malformation, both intracranial and extracranial. These abnormalities are known to exist in the presence of myelomeningocele, and thus, when observed, the fetus should not be considered to have “multiple anomalies” ( Table 9-4 ).
| Anomaly | Frequency |
|---|---|
| Myelomeningocele | Always |
| Hydrocephalus | Almost always |
| Dysplastic tentorium | Almost always |
| Small posterior fossa | Almost always |
| Luckenschädel | Almost always |
| Caudal displacement of brainstem | Usually |
| Cervicomedullary kink | Usually |
| Upward cerebellar herniation | Usually |
| Large massa intermedia | Usually |
| Elongated cranial nerves | Usually |
| Tectal beak | Usually |
| Callosal hypogenesis | Usually |
| Syringohydromyelia | ~50% |
| Malformations of cortical development | Occasionally |
| Aqueductal stenosis | Occasionally |
There is a correlation between the location (vertebral level) and extension of the spinal lesion and the neurologic outcome. In general, lower and smaller defects result in less severe neurologic compromise. The level of the bony spinal lesion can be determined by counting the affected vertebrae either from the most distal vertebra (in the second trimester fetus, the lowest ossified vertebra is S4) or from the rib cage (which ends at T12). Antenatal ultrasound assessment correlates reasonably well with postnatal assessment (75%), and MRI has been found to add little to determination of the bony level of the defect. However, it is not possible to precisely predict motor function, morbidity, or developmental milestones. Of note, the degree of ventricular enlargement does correlate with the need for postnatal shunting.
Antenatal counseling after detection of fetal spina bifida is complex and should involve a multidisciplinary team. Postnatal hydrocephalus requiring shunting, causing incontinence, and resulting in motor weakness requiring wheelchair use is common. Options for management of an affected pregnancy include termination, expectant management with postnatal repair, and in utero surgical repair (see Chapter 24 ). The benefit of cesarean delivery to improve neurologic outcome of infants with open spina bifida is uncertain.
Closed spina bifida is characterized by a vertebral defect (schisis) covered by skin. Most defects are small, involving only a few vertebral segments, and the classic intracranial signs (lemon-shaped calvarium, banana-shaped cerebellum, ventriculomegaly) are absent ( Fig. 9-8 ). As a consequence, the sonographic diagnosis of closed spina bifida is difficult and in practice is only possible in those cases that are associated with a subcutaneous mass, such as a meningocele or lipoma overlying the bony defect. The outcome of closed spina bifida is difficult to predict. These infants do not develop Arnold-Chiari malformation or hydrocephalus. However, those with subcutaneous masses may suffer from neurologic sequelae, including weakness or paralysis of the lower extremities and incontinence, usually as a consequence of tethering or compression of the spinal cord.
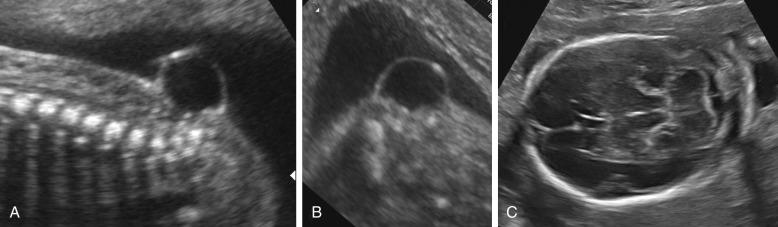
The term cephalocele is used to describe a protrusion of intracranial contents through a bony defect of the skull. Most commonly, these lesions arise from the midline, in the occipital area; they arise less commonly from the parietal or frontal bones. Encephaloceles are characterized by the presence of brain tissue in the lesion. When only meninges protrude, the term cranial meningocele is used. Cephaloceles often cause impaired cerebrospinal fluid circulation and hydrocephalus, and massive encephaloceles may be associated with microcephaly. Associated anomalies are frequent ( Table 9-5 ). Fetal cephaloceles are suspected when a paracranial mass is seen on sonography ( Fig. 9-9 ). The diagnosis of encephalocele is typically straightforward, as the presence of brain tissue inside the sac is striking on ultrasound images, although distinguishing a cranial meningocele from soft tissue edema or a cystic hygroma of the neck may be difficult. The diagnosis of cephalocele is favored when it is possible to demonstrate a bony defect in the cranial vault or associated cerebral abnormalities, such as ventriculomegaly. The pediatric literature suggests that the outcome of cephaloceles is primarily related to the presence or absence of brain tissue inside the lesion. The largest available antenatal series reports a dismal prognosis for both varieties, with the possible exception of small lesions associated with normal intracranial anatomy.
|
Midline Anomalies
Midline cerebral anomalies include a group of brain defects that encompass a wide spectrum of severity and are often associated with craniofacial malformations.
The holoprosencephalies are complex abnormalities of the forebrain characterized by incomplete separation of the cerebral hemispheres and formation of diencephalic structures. They are rarely seen at birth owing to a high intrauterine mortality rate, although these disorders are relatively common in series of antenatally detected CNS anomalies. The cause of holoprosencephaly is heterogeneous, and in most cases, the anomaly is isolated and sporadic. However, holoprosencephaly may also be associated with chromosomal abnormalities (trisomy 13 and polyploidy), a number of single gene disorders, and other anatomic abnormalities.
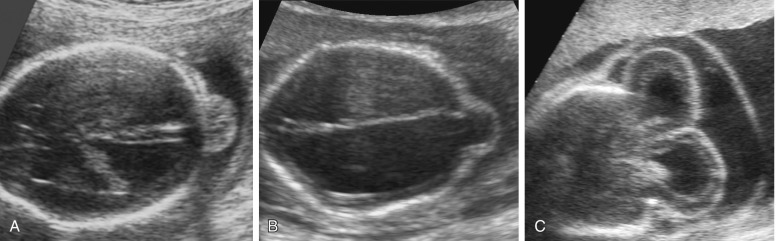
The most widely accepted classification of these disorders recognizes three major varieties: the alobar, semilobar, and lobar types. In addition, a middle interhemispheric variant is described ( Fig. 9-10 ).
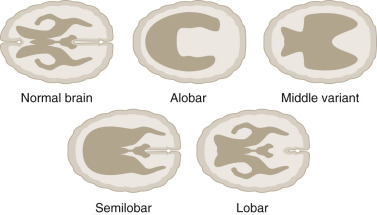
The alobar, semilobar, and middle interhemispheric variant types have a similar appearance on antenatal ultrasound imaging: there is no midline echo anteriorly (absent septum pellucidum), and a single ventricular cavity is present ( Fig. 9-11 ). In the alobar form, which is the most severe, the interhemispheric fissure, falx cerebri, and corpus callosum are absent; there is a single primitive ventricle; the thalami are fused in the midline; and there is absence of the third ventricle, neurohypophysis, and the olfactory bulbs and tracts. In semilobar holoprosencephaly, the two cerebral hemispheres are partially separated posteriorly but there is still a single ventricular cavity. The splenium of the corpus callosum is present without the genu or body of the corpus callosum. In both alobar and semilobar holoprosencephaly, the roof of the ventricular cavity, the tela choroidea (which is normally enfolded within the brain), may balloon out between the cerebral convexity and the skull to form a cyst of variable size called the dorsal sac. Both of these forms are often associated with microcephaly, though they can sometimes be associated with macrocephaly because of a dorsal cyst or obstructive hydrocephalus.
In lobar holoprosencephaly, the interhemispheric fissure is well developed posteriorly and anteriorly, but there is a variable degree of fusion of the cingulate gyrus and the lateral ventricles, and the septum pellucidum is absent. In the middle hemispheric variant, fusion occurs primarily at the level of the bodies of the lateral ventricles, whereas the frontal horns and posterior horns are relatively well developed.
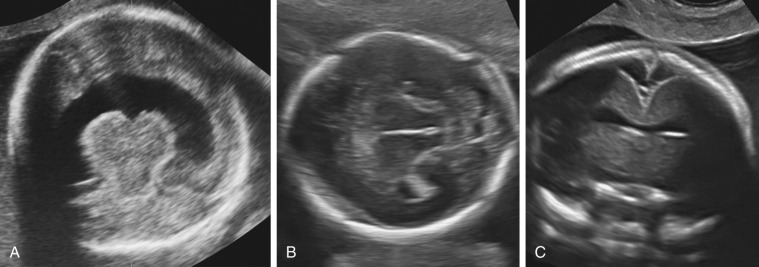
Gross facial abnormalities (cyclopia, hypotelorism, and absence of the nose) are frequently associated with alobar and semilobar holoprosencephaly ( Fig. 9-12 ). Facial malformations include cyclopia and severe hypotelorism with median cleft lip/palate. The nose may be absent, replaced by a proboscis, or flattened. Facial anomalies are rarely associated with lobar holoprosencephaly or the middle interhemispheric variant.
Prenatal diagnosis of alobar or semilobar holoprosencephaly depends on the demonstration of a single rudimentary cerebral ventricle, which may protrude posteriorly through the incompletely enfolded cortex to form a dorsal sac. Additional findings of typical facial anomalies can assist in confirming the diagnosis. In most cases, the diagnosis is possible at 11 to 13 weeks’ gestation. The lobar variety is associated with more subtle findings (absence of the septum pellucidum with central fusion of the frontal horns), and this diagnosis is rarely made prior to 20 weeks’ gestation ( Fig. 9-13 ).
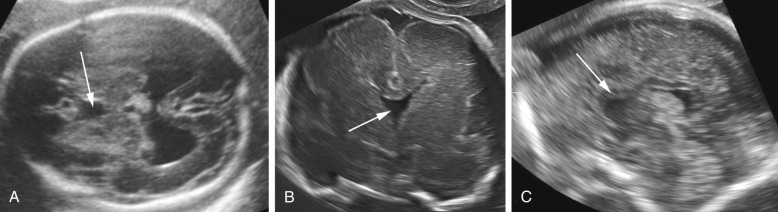
Infants affected by alobar and semilobar holoprosencephaly have an invariably poor prognosis. Antenatally diagnosed lobar holoprosencephaly is also associated with poor postnatal neurodevelopmental outcomes.
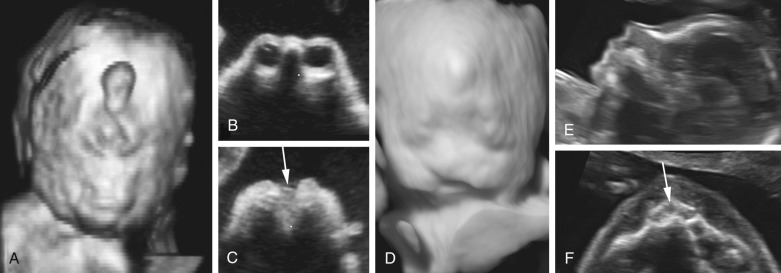
ACC is an anomaly of uncertain prevalence and variable clinical significance. The best estimates suggest a prevalence of approximately 1.4 per 10,000 live births in the general population and 2% to 3% in the developmentally disabled. The cause of ACC is heterogeneous. Genetic causes are common, and the anomaly is associated with a number of different genetic syndromes. The high frequency of associated malformations and chromosomal aberrations suggests that ACC is often part of a widespread developmental disturbance. ACC may be either complete or partial. In the latter case, also referred to as dysgenesis or partial agenesis, the caudad or posterior portion is missing to varying degrees. Hypoplasia refers to a corpus callosum of normal length but with much decreased thickness; this is rarely identified by prenatal sonography.
The diagnosis of ACC is possible from midgestation onward but can be challenging even for expert sonologists. The anatomic elements useful for reaching this diagnosis are summarized in Figure 9-14 .
Related posts:
 Ultrasound Evaluation of Fetal Aneuploidy in the First and Second Trimesters
Ultrasound Evaluation of Fetal Aneuploidy in the First and Second Trimesters
 Measurements Frequently Used to Estimate Gestational Age and Fetal Biometry
Measurements Frequently Used to Estimate Gestational Age and Fetal Biometry
 Role of Doppler Sonography in Obstetrics
Medications and Reported Associated Malformations
Role of Doppler Sonography in Obstetrics
Medications and Reported Associated Malformations
 Role of Sonography in Gynecologic Interventions
Role of Sonography in Gynecologic Interventions
 Ultrasound Features of Fetal Syndromes
Ultrasound Features of Fetal Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


