Up to 8% of renal cancers are thought to have a hereditary component. Several hereditary renal cancer syndromes have been identified over the last few decades. It is important for the radiologist to be aware of findings associated with hereditary renal cancer syndromes to detect tumors early, enroll patients in appropriate surveillance programs, and improve outcomes for the patient and affected family members. This review discusses from a radiologist’s perspective well-known hereditary renal cancer syndromes and emerging genetic mutations associated with renal cancer that are less well characterized, focusing on imaging features and known associations.
Key points
- •
Hereditary renal tumors are diagnosed at a younger age than sporadic tumors and are often multiple and multifocal.
- •
All known subtypes of renal cancer have been identified in hereditary syndromes.
- •
Most hereditary cancers are low grade and therefore less aggressive. However, some hereditary renal cancers are highly aggressive.
- •
Radiologist awareness of extrarenal findings associated with hereditary renal cancer syndromes is helpful for early diagnosis.
Introduction
Renal cell carcinoma is the eighth most common type of cancer, with an estimated 73,820 new diagnoses in 2019 in the United States. About one-fifth of patients with renal cell carcinoma have metastases at the time of presentation. Hereditary renal cancers (HRNs) account for an estimated 5% to 8% of renal cancers, although this may be an underestimate because of genetic factors not yet characterized. The diagnosis of a hereditary renal tumor has important implications for the patient and their family. For the patient, knowledge of a germline mutation can play a pivotal role in the future management and treatment of the cancer. Patients with a possible hereditary component to their renal cancer should be referred for genetic testing, which can help guide disease management and inform whether family members could be at risk. Patients with a family history, unique personal history, bilateral or multifocal disease, or early age at diagnosis are at increased risk and therefore, may benefit from genetic testing. Recognition of clinical and imaging features associated with hereditary renal carcinoma is an important part of diagnosis. Imaging findings outside the kidneys may be the first indication of a possible hereditary cancer syndrome. Therefore, knowledge of not only renal tumors but other visceral manifestations of hereditary cancer syndromes is important for a radiologist to have, to recognize HRC syndromes.
Several known genetic mutations are associated with HRC. There are mutations that cause well-characterized renal-epithelial syndromes including von Hippel-Lindau (VHL) syndrome, Birt-Hogg-Dube syndrome (BHD), hereditary leiomyomatosis and renal cell cancer syndrome (HLRCC), tuberous sclerosis complex (TSC), and hereditary nonpolyposis colorectal cancer syndrome (HNPCC). Other genetic mutations associated with HRC are emerging but are less well characterized, including chromosome 3 translocations, BAP1 cancer syndrome, PTEN mutation (Cowden syndrome), succinate dehydrogenase complex subunit B (SDHB), MITF predisposition, and CDCJ3 (hyperparathyroidism jaw tumor syndrome). HRCs are described by their frequency, type of tumor produced, treatments, and aggressiveness among others. This review discusses HRCs from a radiologist’s perspective, focusing on imaging features and known associations.
von Hippel-Lindau syndrome
VHL syndrome has a frequency of approximately 1:36,000, making it the most common identified cause of HRC. VHL is an autosomal-dominant hereditary disease caused by mutations in the VHL tumor suppressor gene. There is 90% penetrance by age 60. Nonetheless, up to 20% of cases are caused by sporadic mutations first occurring in the index patient with no antecedent family history. , Nonrenal manifestations include pancreatic neuroendocrine tumors (pNET), pancreatic cysts, central nervous system (CNS) hemangioblastomas, endolymphatic sac tumors, retinal angiomas, pheochromocytomas, epididymal papillary cystadenomas, and cystadenomas of the broad ligament. Clear cell renal cell cancer (ccRCC) is the main malignant neoplasm in VHL affecting 24% to 45% of patients. Additionally, of these findings, ccRCC has historically been associated with the highest mortality, but because of improved surveillance and earlier diagnosis ccRCC mortality has been superseded by VHL-related CNS diseases. , Tumor cells lacking a functional VHL tumor suppressor gene overproduce the downstream products of hypoxia-inducible factor target genes, such as vascular endothelial growth factor. This predisposes patients to developing hypervascular masses in various organs.
Renal Imaging Findings
The renal cancer type associated with VHL is virtually always ccRCC, although the precise pathogenesis is not completely understood. Most frequently, it manifests as a hyperplastic or metaplastic clear cell lining of multiple renal cysts from which ccRCC can develop. Later in the progression, solid and cystic components are also seen. All renal cysts in a patient with VHL should be considered premalignant and followed by imaging, although variable behavior is expected, including spontaneous involution.
On computed tomography (CT), lesions have a water attenuation initially because of their cystic nature. On MR imaging, lesions are T1-hypointense and T2-hyperintense. Fat-suppressed T1-weighted MR imaging with contrast can help to differentiate complex cystic components from solid enhancing components. Lesions are characterized as mostly cystic or mostly solid-enhancing, because they are commonly heterogeneous ( Fig. 1 ). ccRCC progression is marked by enlarging solid components, enhancing septations, and solid nodules. As solid components grow, they take over cystic components until the renal masses are comprised of nearly all solid tumor. Because of this, the amount of solid rather than cystic disease determines which lesions warrant treatment.
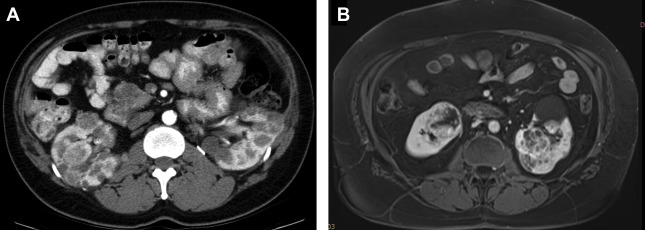
Solid components of renal masses often demonstrate avid enhancement on contrast-enhanced imaging. The clear cell subtype of RCC shows stronger enhancement in the corticomedullary phase of contrast media enhancement than other tumor subtypes and has more persistent enhancement in the nephrogenic and excretory phase. MR imaging can be substituted for CT in detecting VHL tumors and is superior to ultrasound. MR imaging reduces the radiation burden to patients undergoing multiple studies over their life. VHL tumors are generally T2 bright and T1 hypointense with avid enhancement on different phases of postcontrast T1 similar to CT. The VHL tumors are generally low grade, and demonstrate higher mean apparent diffusion coefficient values than other more aggressive subtypes, such as papillary and chromophobe RCC. ,
Extrarenal Manifestations
In addition to renal lesions, abdominal imaging studies may reveal pancreatic cysts, serous cystadenomas of the pancreas, and solid enhancing nonfunctional pNET. Pancreatic lesions are often multiple and are distributed throughout the pancreas, although solid pNET tumors typically are more concentrated in the head and uncinate process. The most common histologic subtype of cystic pancreatic lesions in VHL is serous cystadenoma that typically has a benign course, although pancreatic insufficiency may be seen. On imaging, pancreatic cystadenomas typically display a central scar with cysts arranged in a rosette pattern around it. Calcification is frequently seen. Serous cystadenomas can grow larger but are not malignant. Patients with VHL with pNET are more likely to have missense mutations in the VHL gene and can develop metastatic disease. An association between pNET metastasis and exon 3 mutations in patients with VHL has been suggested but further studies are needed. On CT they are typically highly vascular and are detected during the arterial phase of contrast administration. Fluorodeoxyglucose (FDG)-PET/CT has been shown to increase sensitivity for detecting pNET over anatomic imaging. 68Ga-Dotatate PET/CT had a significantly higher detection rate for pNET when compared with anatomic imaging for all lesions, and comparable detection rate for pancreatic lesions in patients with VHL. When small, pNET are well circumscribed and hypervascular and often found in the uncinate process. Larger pNETs may become necrotic and appear more heterogeneous. These tumors are bounded by a capsule and thus, displace rather than invade surrounding structures. However, as the tumor grows its margins may become less distinct.
In men, VHL is characterized by multiple hyperenhancing papillary cystadenomas in the epididymis, occurring in up to 40% of patients, whereas in women cystadenomas may be present in the broad ligament of the uterus. In the adrenals and paraganglia, pheochromocytomas occur. These are bilateral and/or multifocal, subclinical or highly symptomatic. In addition to biochemical evidence of excess catecholamine production, abdominal CT and MR imaging with contrast are sensitive in detecting adrenal and extra-adrenal pheochromocytomas in these patients. These lesions are typically hypervascular. Paragangliomas (extrarenal pheochromocytomas) can vary in appearance on cross-sectional imaging but typically show strong arterial enhancement. If rapidly growing, a necrotic core may be present. The presence of multiple paragangliomas suggests a particular subtype of VHL called type 2 VHL, which is associated with point mutations in the VHL gene.
Within the CNS, the most common lesion is the hemangioblastoma, typically located in the posterior fossa of the brain. On CT or MR imaging these lesions demonstrate avidly enhancing solid and cystic components. Hemangioblastomas in the CNS typically present as mural enhancing nodules on MR imaging with adjacent cystic spaces and can lead to multiple symptoms, particularly difficulty with balance. A similar lesion is found in the retina where they are named retinal capillary angiomas. Diagnosing these retinal lesions early can prevent blindness. This is done with careful eye examinations, and often they are the first manifestation of VHL. Patients with VHL also may develop endolymphatic sac tumors in the inner ear, which can cause deafness if left untreated. They have the potential to invade surrounding bone and infiltrate into the extradural space. On T2-weighted MR imaging, endolymphatic sac tumors are heterogeneous hyperintense masses with avid enhancement.
Screening and Management
Lifelong surveillance is necessary in patients with VHL given the ongoing risks for tumor development with increasing age. Longitudinal surveillance has been shown to decrease morbidity and mortality. Multiple groups have developed surveillance guidelines. The most recent comes from the VHL Alliance last updated in 2017, which recommends screening for retinal hemangioblastoma with ophthalmoscopy starting in infancy because of the noninvasive nature of the examination. Starting at age 5, plasma free metanephrines or 24-hour urinary fractionated metanephrines should be checked annually. In families with a strong history of pheochromocytoma/paraganglioma, this is initiated in patients at 2 years old. Patients with VHL should be screened for RCC, pheochromocytoma, and pNET with abdominal ultrasound annually up to age 16, and then yearly thereafter with alternating ultrasound/MR imaging gradually switching to MR imaging in adulthood. CNS hemangioblastomas should be screened for with biennial MR imaging of the brain and spine starting at age 16, but it may be desirable to begin MR imaging screening as soon as the patient is old enough to tolerate MR imaging without sedation. Pancreatic cysts, pNETs, renal cancers, and pheochromocytomas should all be followed typically with MR imaging, which avoids the radiation associated with CT. Although pancreatic cysts do not necessitate treatment unless they are symptomatic, pNETs have metastatic potential and should be monitored for growth. Although it was once thought that intravenous contrast could induce hypertensive crisis in patients with paraganglioma and pheochromocytoma, there is no evidence for this and so there is no contraindication. There is some concern in the VHL community regarding the cumulative effect of gadolinium after many years of screening MR imaging with contrast. It is known that gadolinium may accumulate in the brain and other organs, although no adverse neurologic effect has been reported. Using gadolinium contrast agents with strong chemical chelates (ie, macrocyclic agent) reduces the potential risk of gadolinium deposition. Gadolinium enhancement provides useful diagnostic information in many VHL cases so the issue needs to be carefully discussed with concerned patients.
If a patient with VHL is diagnosed with RCC, nephron-sparing surgery is the recommended management for tumors greater than or equal to 3 cm because of the risk of metastasis, whereas smaller lesions are monitored safely with imaging surveillance. Improved imaging modalities have led to the detection of more renal cancers at an early stage. Solid tumors less than 3 cm are thought to have near zero metastatic potential and are safely monitored until they reach the 3 cm threshold for surgery. Partial nephrectomy has been shown to be as effective as total nephrectomy for early stage RCC, and other nephron-sparing approaches, such as cryofrequency and radiofrequency ablation, may be appropriate for small tumors while minimizing damage to healthy parts of kidney. Laparoscopic surgery has greatly reduced the recovery period from treatment.
Birt-Hogg-Dube syndrome
BHD syndrome is an autosomal-dominant condition caused by an inactivating germline mutation in the folliculin (FLCN) tumor suppressor gene. The syndrome presents with lung cysts; pneumothorax; fibrofolliculomas of the skin; and the most threatening potential complication, RCC. Although the penetrance of renal cancer is low, patients with BHD have up to seven times the risk of renal cancer compared with the general population. Approximately 13% to 34% of patients have renal tumors, but unlike VHL, the tumors vary in histologic subtype with the most common being oncocytic-chromophobe type. Major criteria for BHD are greater than five cutaneous fibrofolliculomas or a confirmed germline mutation in FLCN. Minor criteria include family history of BHD, early onset or multifocal renal cancer, and lung cysts. One major or two minor criteria establish a diagnosis.
Renal Imaging Findings
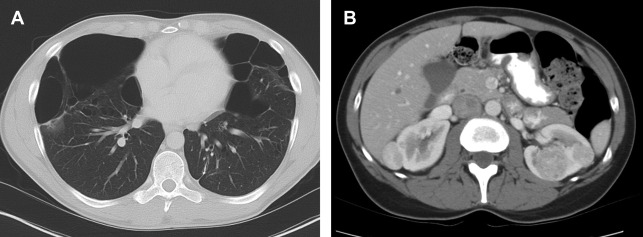
Renal tumors in BHD syndrome are noted at an earlier age than sporadic RCC, with an average age of 50.4 years. The tumors range in histology from oncocytomas (benign) to chromophobe carcinomas (malignant). Oncocytic-chromophobe renal cell carcinoma tend to exhibit homogenous enhancement on CT and MR imaging with scant if any cystic components distinguishing them from VHL ( Fig. 2 ). Central necrosis is not usually seen, but calcifications are common, and the tumors are notable for their uniformity and absence of radiographic necrosis. Compared with other hereditary RCC syndromes, the tumors associated with BHD are somewhat less aggressive and can be monitored for longer; however, aggressive tumors have been documented. ,
Extrarenal Manifestations
Lung cysts affect more than 80% of patients with BHD and are usually multiple and found predominantly in the lower lobes (see Fig. 2 ). The cysts can grow to be >7 cm, and are frequently multiseptated when large. Spontaneous pneumothoraxes secondary to pulmonary cysts are commonly seen, occurring in 23% to 30% of patients younger than age 40. Cutaneous findings affect 90% of patients and include fibrofolliculomas, trichodiscomas, and acrochordons. Additional cancers that may be associated with the FLCN suppressor gene mutation include clear cell thyroid carcinoma and colorectal carcinoma. ,
Screening and Management
Surveillance is recommended for these patients, but the optimal imaging method and interval remain to be defined. It has been proposed that all patients should undergo high-resolution CT of the chest and MR imaging of the abdomen at diagnosis. At-risk BHD family members should also undergo baseline surveillance with abdominal MR imaging. Screening for renal tumors is recommended starting at age 20, either annually or biannually with MR imaging. When the largest tumor reaches 3 cm, surgical intervention, typically laparoscopic partial nephrectomy, is recommended. Nephron-sparing surgery to preserve renal function is preferred, because patients with BHD may develop more tumors and require repeated surgeries during their lifetime. ,
Tuberous sclerosis complex
TSC, also historically known as Bourneville-Pringle disease, affects an estimated 1 million people worldwide with a frequency of 1:10,000. TSC is caused by germline mutations in TSC1 or TSC2, which encode hamartin and tuberin, respectively. Mutations in TSC2 result in more severe disease manifestations. TSC is acquired as a spontaneous mutation, which occurs in approximately 70% of cases, or by hereditary autosomal-dominant transmission accounting for the remainder. The absence of family history in most cases can make early diagnosis more challenging. TSC is characterized by the presence of multiple hamartomas, primarily on the skin and in the CNS. The risk of renal cancer in patients with TSC is estimated to be 1% to 4%, , but 70% to 90% of patients with TSC develop one or more benign renal angiomyolipomas (AMLs). Although they have no malignant potential, AMLs are at risk for spontaneous hemorrhage when they grow larger than 4 cm in diameter and may need to be treated with transarterial embolization.
Renal Imaging Findings
AMLs are by far the most common lesion in the kidney in patients with TSC. AMLs are a benign neoplasm composed of dysmorphic blood vessels, smooth muscle, and adipose tissue. Typical AMLs involve the renal cortex and have a macroscopic fat component ( Fig. 3 ), but AMLs without gross visible fat occur in approximately one-third of patients with TSC and can mimic cancer. The AMLs without gross fat demonstrate higher attenuation than normal renal parenchyma and enhance homogenously. AMLs can also mimic cancer by extending into local lymph nodes and the inferior vena cava. The next most common renal lesions in TSC are benign renal cysts, which occur in up to 47% of patients. The cysts are not considered premalignant as they are in VHL.
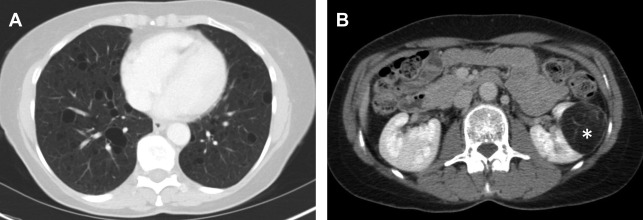
Although there are a plethora of nonmalignant findings associated with TSC that can help a radiologist identify it, the only cancer patients with TSC are at increased for is RCC and even then, the risk is low. Clear-cell, chromophobe, or papillary subtypes of RCC have all been associated with TSC. Differentiating renal cell carcinoma from an AML without gross fat is often challenging, but MR imaging is useful in differentiating the two. Intratumoral necrosis and larger size are associated with RCC, whereas low SI index on T2-weighted images relative to renal parenchyma and smaller size are associated with AML. Specifically, it has been found that T2 SI ratio less than 0.9 or an arterial/delayed enhancement ratio greater than 1.5 favor a diagnosis of AML with 99% sensitivity and 73% specificity. AMLs without gross fat also tend to enhance more uniformly than RCC. Despite these suggestive findings, biopsy of the lesion may be desirable before any treatment is contemplated.
Extrarenal Manifestations
TSC is characterized by benign hamartomas, especially in the brain. Ninety percent of patients are affected by subependymal nodules and cortical tubers. They are multiple and can result in neurologic manifestations, such as epilepsy and cognitive disability. On CT of the head, subependymal nodules are seen along ventricular walls, often appearing as calcified foci. On MR imaging, they are isointense to hyperintense on T1- and T2-weighted images. In the frontal lobes, cortical tubers are hypointense lesions on T1-weighted images, and hyperintense on T2 and FLAIR images. Unlike cortical tubers, cerebellar tubers are wedge-shaped and not epileptogenic. Giant cell astrocytomas, a larger variant of subependymal tubers, affect 5% to 15% of patients with TSC and can cause hydrocephalus because of their rapid growth and location adjacent to the interventricular foramen of Monro. They are heterogeneous on CT and MR imaging and may have intratumoral hemorrhage and calcification. Other less common extrarenal manifestations include rectal polyps, bone islands, thyroid adenomas, and lymphangioleiomyomatosis (LAM). LAM, affecting 26% to 49% of women with TSC, causes cystic lung destruction and potential pneumothorax and chylous pleural effusions (see Fig. 3 ). Cardiac rhabdomyomas occur in 50% to 70% of infants with TSC but usually spontaneously resolve in the perinatal period.
Screening and Management
There are limited guidelines available for screening and management of this disease because of a lack of available evidence and low risk. Because of antenatal detection of cardiac rhabdomyomas on ultrasound, the median age at diagnosis is only 6 months. In general, CNS involvement and seizures followed by dermatologic manifestations are the leading features at presentation. LAM has a reported prevalence of 27% in females with TSC aged less than 21 years, thus it is recommended to begin screening with high-resolution CT at age 18 in females with TSC. , LAM is treated pharmacologically with mTORC1 inhibitors, such as sirolimus, which is Food and Drug Administration approved for the treatment of LAM. Imaging surveillance with abdominal MR imaging or CT is recommended at least every 3 years, although yearly abdominal MR imaging has been recommended by several groups mainly to monitor AML growth. , Renal imaging is typically dictated by the presence of AMLs but radiologists should remain alert for malignant lesions. Once renal cancers reach about 3 cm in diameter, surgical intervention is recommended, typically nephron sparing or by focal ablation. Renal function should be monitored in patients who undergo repeat surgeries.
Hereditary papillary renal cancer
Hereditary papillary renal cancer (HPRC) is a rare familial cancer syndrome (approximate incidence of 1 in 10 million) caused by an activating mutation in the MET protooncogene. It is an autosomal-dominant disorder with high penetrance. HPRC is associated with an increased risk of type 1 papillary renal carcinoma, with two-thirds of patients developing the cancer by age 60% and 90% by age 80. , Type 1 papillary renal carcinoma is less aggressive than type 2, and patients can often be placed on active surveillance for their renal lesions. Unlike other HRC syndromes, the only known manifestation of this disease is papillary renal cancer. Renal tumors associated with HPRC generally appear after the age of 30, but an early onset phenotype has been identified that presents in the second decade of life. The disease is limited to a small number of family clusters.
Renal Imaging Findings
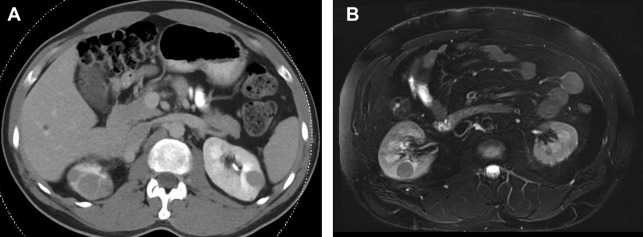
HPRC produces multiple bilateral tumors that may number in the hundreds or even thousands, most of which are microscopic. Type I papillary tumors tend to be slow growing and often do not present until later in life. The lesions are hypovascular with minimal enhancement on MR imaging and CT. Enhancement may be so low that it is difficult to qualitatively differentiate the lesions from cysts. For this reason, a quantitative cutoff of 10 to 30 HU increase on contrast-enhanced CT or 15% increase in signal intensity on T2-weighted MR imaging has been previously defined. Ultrasound is not recommended for detection or follow-up of renal lesions in HPRC because they are isoechoic with background and difficult to see. However, ultrasound is used to confirm that a poorly enhancing lesion on CT or MR imaging is solid and not cystic. MR imaging is the most accurate modality in differentiating papillary RCC from other types of RCC with low enhancement, T2 signal hypointensity, and homogenous restricted diffusion on diffusion-weighed imaging ( Fig. 4 ). ,
Related posts:
 Image Interpretation
Image Interpretation
 Protocol Optimization for Renal Mass Detection and Characterization
Protocol Optimization for Renal Mass Detection and Characterization
 Radiomics and Artificial Intelligence for Renal Mass Characterization
Radiomics and Artificial Intelligence for Renal Mass Characterization
 Percutaneous Thermal Ablation for Treatment of T1a Renal Cell Carcinomas
Percutaneous Thermal Ablation for Treatment of T1a Renal Cell Carcinomas
 Imaging of Renal Infections and Inflammatory Disease
Imaging of Renal Infections and Inflammatory Disease
 Use of Contrast Ultrasound for Renal Mass Evaluation
Use of Contrast Ultrasound for Renal Mass Evaluation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


