Etiology
Urinary tract anomalies encompass a wide range of abnormalities from the multiple varied components of the urinary tract—the renal parenchyma, the collecting system, the bladder, the urethra, and the vasculature. Anomalies result from alterations in the normal embryologic development of the urinary system. Detecting these anomalies requires an understanding of the embryologic development of the urinary system.
Prevalence and Epidemiology
Overall, it is estimated that congenital anomalies of the kidney and urinary tract occur in approximately 1 in every 500 newborns and account for 20% to 30% of all anomalies in the prenatal period. Many of these anomalies manifest during fetal development, carrying risks to the fetus, and are detected at routine ultrasound screening during pregnancy. However, some are detected decades later as physical ailments related to the urinary system or incidentally during imaging performed for other purposes. Congenital anomalies of the urinary tract are a major cause of renal failure in infants and a cause of hypertension and cardiovascular disease in adults.
Prevalence and Epidemiology
Overall, it is estimated that congenital anomalies of the kidney and urinary tract occur in approximately 1 in every 500 newborns and account for 20% to 30% of all anomalies in the prenatal period. Many of these anomalies manifest during fetal development, carrying risks to the fetus, and are detected at routine ultrasound screening during pregnancy. However, some are detected decades later as physical ailments related to the urinary system or incidentally during imaging performed for other purposes. Congenital anomalies of the urinary tract are a major cause of renal failure in infants and a cause of hypertension and cardiovascular disease in adults.
Normal Anatomy
Urinary tract development begins at the 4th gestational week when the intermediate mesoderm separates into three parts: pronephros, mesonephros, and metanephros. In the cervical region these nephrotomes form the pronephros, a rudimentary urinary system that regresses before birth. The second segmented portion becomes the mesonephros, which gives rise to the ureteral bud that becomes the renal collecting system. The third segmented portion is the metanephros, which becomes the permanent kidney.
At the 5th week of gestation, the ureteral bud, arising from the mesonephros, penetrates the adjacent metanephros and dilates, forming the renal pelvis. Subsequently, it divides into cranial and caudal portions, becoming the major calyces. These calyces continue to subdivide, forming the minor calyces, the renal pyramids, and the 1 to 3 million collecting tubules.
The metanephros, stimulated by the penetration of the ureteral bud, develops into nephrons. Portions of these nephrons lengthen and become the proximal convoluted tubules, the loop of Henle, and the distal convoluted tubules. The metanephric tissue eventually migrates craniad and is positioned in the retroperitoneal space in the upper lumbar native position of the kidneys.
The distal portion of the mesodermal tissue becomes the posterior wall of the bladder, including the trigone and bladder neck. The ventral cloaca develops into the urogenital sinus, which eventually becomes the remaining portions of the bladder and urethra.
During embryologic development, multiple renal arteries are created as lateral intersegmental arteries arising from the mesonephros. As the kidneys migrate craniad, the lower arteries regress. The final, definitive renal artery comes from the artery originally supplying the adrenal glands.
Specific Lesions
To organize the numerous and varied congenital anomalies of the urinary tract, the entities have been organized under subcomponents of the urinary tract system: the renal parenchyma, collecting system, bladder, urethra, and renal vasculature.
Renal Parenchyma
Anomalies of the renal parenchyma are organized into the following major categories: renal agenesis, renal hypoplasia, renal dysmorphisms, anomalies of renal ascent, and renal fusion anomalies.
Renal Agenesis
Unilateral regional agenesis occurs in approximately 1 in 5000 newborns. Bilateral renal agenesis occurs in 1 in 30,000 newborns.
Pathophysiology.
On a molecular scale, renal agenesis has been linked to the failure of glial-derived neurotropic factor. Embryologically, renal agenesis occurs because of early degeneration of the ureteral bud, which fails to reach the metanephric tissue cap. Bilateral regional agenesis appears to have a genetic cause and is twice as common in males as in females.
Clinical Presentation.
Bilateral renal agenesis is rare. Bilateral renal agenesis was described by Edith Potter in 1946 and is a component of Potter’s syndrome, in which there is oligohydramnios or anhydramnios (absent amniotic fluid) owing to a renal developmental abnormality. It is suspected clinically with oligohydramnios as a result of inability for the fetus to excrete the swallowed amniotic fluid. Although the fetus will survive gestation because the kidneys are not necessary for exchange of waste products, bilateral renal agenesis is incompatible with life after birth and typically the neonate will die within a few days. Unilateral renal agenesis may remain undetected if there is normal renal function.
Imaging.
Bilateral renal agenesis is detected on screening pregnancy ultrasonography as oligohydramnios and nonvisualization of the kidneys. Unilateral renal agenesis is visualized as absence of one kidney in either the native retroperitoneal location or in the pelvis.
Unilateral renal agenesis typically is incidentally visualized on computed tomography (CT) or magnetic resonance imaging (MRI) as absence of one kidney ( Figure 69-1 ). Often there is compensatory enlargement of the solitary kidney.
Treatment.
Bilateral renal agenesis is incompatible with life. Unilateral renal agenesis requires no treatment owing to compensatory hypertrophy of the solitary kidney. In patients who have poor renal function, dialysis may be necessary.
Renal Hypoplasia
In the U.S. Renal Data System all hypoplasias and dysplasias, reported in a single category, account for 8.9% of pediatric patients presenting with end-stage renal disease. The size of the kidneys depends on the number of nephrons created during embryologic development. The average number of nephrons ranges from 300,000 to 1 million in each kidney.
Pathophysiology.
The most common cause of renal hypoplasia is defective ureteral branching in embryologic life. Other factors implicated include nutrition during pregnancy and genetic factors.
Clinical Presentation.
There may be no clinical manifestations of the disease, and renal hypoplasia may be detected incidentally as a decrease in the size of one or both kidneys. However, patients with renal hypoplasia are at risk for developing primary hypertension. These patients are reported to have 46% fewer glomeruli than normal individuals and are at risk for chronic renal failure. Patients can present with anorexia, vomiting, and failure to thrive and also with short stature, polyuria, polydipsia, and proteinuria.
Imaging.
On ultrasonography, CT, or MRI, renal hypoplasia is visualized as a decrease in the size of kidneys.
Treatment.
There is no specific treatment. Patients with renal failure may consider dialysis. Primary hypertension is treated with medication.
Renal Dysmorphisms
Renal dysmorphisms include a wide variation of anomalies of the renal cortical contour. Documented dysmorphisms include persistent fetal lobulation, junctional parenchymal defect, dromedary hump, septum of Bertin, aberrant papilla, and sinus lipomatosis. Renal dysmorphisms occur secondary to embryologic developmental anomalies of the metanephros. Many of these dysmorphisms are seen as variants in the appearance of the kidney.
Persistent Fetal Lobulation.
Persistent fetal lobulation is the result of fetal lobulation that persists into adulthood. Typically, the fetal kidneys are subdivided into lobes by grooves that disappear by the end of the fetal period. This anomaly is discovered incidentally and carries no clinical significance. It is important in imaging to distinguish between lobulation and scarring, which can occur from reflux or chronic infection. Lobulation can be seen on CT or ultrasonography as indentations that occur between the medullary pyramids ( Figure 69-2 ).
Junctional Parenchymal Defect.
Junctional parenchymal defect is a fusion defect in the upper pole of the kidney. It is the result of partial fusion of the embryonic renal parenchyma. The incomplete fusion occurs at the junction of the fusing parenchyma. This disorder has no clinical significance. It can be confused with a renal scar or laceration. It typically appears as an echogenic linear defect on ultrasound that extends from the renal sinuses to the cortex ( Figure 69-3, A ). On CT it is seen as a defect extending to the renal sinus (see Figure 69-3, B ).
Dromedary Hump.
Dromedary hump represents prominence of the cortical parenchyma, typically of the left kidney, owing to the compression from the adjacent spleen. It has no clinical significance and should not be confused with a solid renal tumor. On ultrasonography, CT, and MRI, dromedary hump is seen as a focal bulge arising from the lateral cortex of the left kidney ( Figure 69-4 ).
Septum of Bertin.
The septum of Bertin is a normal variant that represents hypertrophied cortical tissue between the medullary pyramids that projects into the renal sinus. It carries no clinical significance. However, it can occasionally be confused with a solid renal tumor.
Septum of Bertin often occurs in the middle third of the kidney and more commonly on the left. On ultrasonography it is seen as cortical tissue indenting the renal sinuses. Although typically similar to the renal cortex in echogenicity, it is also reported to be more echogenic than the renal cortex. On CT and MRI, it is seen as a portion of the cortex indenting into the renal sinuses and enhancing similar to the cortex.
Lobar Dysmorphism.
Lobar dysmorphism represents a pseudotumor in the central kidney resulting from anomalous embryologic development. It is distinguished from the septum of Bertin by the finding of calyx in the central portion of the mass, reflecting a complete renal lobe rather than cortical tissue positioned in the central kidney. This anomaly has no clinical significance and should not be confused with a renal tumor.
On ultrasonography, lobar dysmorphism is seen as a central mass that can be difficult to distinguish from a renal tumor or septum of Bertin. Cortical and nephrographic contrast-enhanced CT can distinguish this central mass from septum of Bertin by the finding of a small central collecting calyx in the mass.
Aberrant Papilla.
An aberrant papilla is an otherwise normal papilla that projects into the lumen of the renal infundibulum or renal pelvis. It results during embryologic development from a shortened minor calyx. This anomaly has no clinical significance and should not be confused with a collecting system tumor.
On excretory phase CT, an aberrant papilla is seen as a small filling defect that projects into the renal calyx ( Figure 69-5 ). It typically has a broad base, and the tip is smooth and conical. It has a similar appearance on intravenous pyelography (IVP).
Sinus Lipomatosis.
Sinus lipomatosis represents an abundance of fat in the renal sinuses. It is typically associated with parenchymal atrophy from severe renal infection or vascular ischemia. It also can be seen in patients who have increased endogenous steroids or who take exogenous steroids. It has no clinical significance and typically does not cause calyceal obstruction. It should be distinguished from fat-containing neoplasms in the renal sinuses.
On imaging, sinus lipomatosis is visualized as increased central renal fat. On ultrasonography it is seen as increased echogenicity, and on CT it is visualized as increased fat density adjacent to the central renal collecting system ( Figure 69-6 ).
Anomalies of Ascent
Pelvic Kidney.
One or both kidneys can be positioned in the pelvis remote from their expected anatomic location in the upper retroperitoneum. The incidence of pelvic kidney is 1 in 725 births.
Pathophysiology.
After the kidney forms in the pelvis from the metanephric tissue, it ascends to a more cranial position in the abdomen because of a decrease in the body curvature as well as growth of the lumbar and sacral regions. Interruptions in this ascent may occur at the level of the arterial fork formed by the umbilical arteries. Failure to pass this area can result in a kidney close to the common iliac artery and consistent with a pelvic kidney. Pelvic kidney is associated with vesicoureteral reflux, hydronephrosis, hypospadias, and contralateral renal agenesis.
Imaging.
On ultrasonography, CT, and MRI there is absence of the kidney in its native upper retroperitoneal location. The kidney can be found in the pelvis and its vascular supply identified arising from the iliac vessels ( Figure 69-7 ). Pelvic kidneys may occasionally be mistaken for pelvic adenopathy.
Renal Fusion Anomalies
Crossed-Fused Ectopia.
Kidneys are positioned both on the same side with crossing of the ureters. Usually they are fused, but they can occasionally be separate. This anomaly occurs more often on the left than on the right. Renal fusion, ectopia, and rotation anomalies occur in 1 in 500 births.
Pathophysiology
Crossed ectopia or crossed-fused ectopia is believed to occur because of abnormal growth of the ureteral bud, resulting in failure of the nephrogenic cell masses to separate.
Clinical Presentation
This disorder may be asymptomatic. Associated clinical findings may not be detected until later in life and include obstructive uropathy, infection, and reflux.
Imaging
Imaging will show both kidneys in the same side of the abdomen ( Figure 69-8 ). They can be fused. Typically, the abnormally placed kidney will have a ureter that crosses to the correct side.
Pancake Kidney.
A pancake kidney is a fusion anomaly in which the upper and lower poles fuse and the resulting fusion is ring-like. It is also known as a disc kidney or doughnut kidney.
Pathophysiology
Pancake kidney is caused by failure of the metanephric tissues to separate.
Clinical Presentation
Most patients have no symptoms. When present, symptoms may be secondary to hydronephrosis, infection, stone formation, or hematuria.
Imaging
CT and MRI can show the fused disc-shaped kidney ( Figure 69-9 ).
Horseshoe Kidney
A horseshoe kidney is fusion of the lower pole of both kidneys. It occurs in 1 in 600 individuals and is slightly more common in females.
Pathophysiology
The kidneys cease embryologic ascent at the root of the inferior mesenteric artery, and the lower poles are pushed together, causing them to fuse.
Clinical Presentation
Clinically, patients may have other congenital anomalies, including cardiac and skeletal disorders and trisomy 18 and Turner’s syndrome. Individuals with horseshoe kidney are at increased risk for developing ureteropelvic junction (UPJ) obstruction and urinary tract stones. There is also a slight increased risk for Wilms’ tumor and carcinoid. The incidence of renal cell carcinoma is the same as for individuals with normal kidneys.
Imaging
On imaging the kidney can be seen to have medially oriented lower poles fused underneath the inferior mesenteric artery ( Figure 69-10 ). The renal pelvis and ureters of both kidneys are positioned anteriorly.
Treatment
Treatment is based on the management of complications of the horseshoe kidney, including surgical management of UPJ obstruction with pyeloureteroplasty and management of stone disease with lithotripsy or surgery.
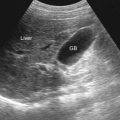
Multicystic Dysplastic Kidneys
Cystic dilation of the collecting tubules can occur in the renal cortex. It is the most common cause of cystic disease in infants and the second most common cause of an abdominal mass in a neonate. It occurs in 1 in 2400 live births and is slightly more common in males. The condition may be unilateral or bilateral and is detected prenatally in almost 90% of cases. When unilateral, the rate of associated malformation or syndrome is 16%. If bilateral multicystic dysplastic kidneys are present, there is a higher rate of associated syndrome or malformation, and only one third of those affected are liveborn.
Pathophysiology
Collecting tubules of the kidneys enlarge into cysts secondary to obstruction or atresia of the ureteral bud before 8 to 10 weeks of fetal life. The anomaly is most commonly unilateral but can be segmental or bilateral. The following two types have been described:
- •
Potter IIa: Multicystic kidney seen as a large kidney with multiple large cysts
- •
Potter IIb: Small kidneys
Clinical Presentation
Clinically, infants can present with a large abdominal mass. If the disorder is bilateral, infants can die of pulmonary hypoplasia. Occasionally, if unilateral, the kidneys can remain undetected until adulthood, at which time they can manifest with intermittent abdominal pain, hematuria, or recurrent urinary tract infections (UTIs).
Imaging
On ultrasonography multiple cysts of various shapes and sizes are seen ( Figure 69-11 ) in Potter IIa disease. The largest cysts are peripherally oriented with replacement of the normal renal architecture. In Potter IIB disease the kidneys are small and echogenic.