Chromosomal aberrations are the most frequent and significant disorders and are detected in five per 1000 live births. The forms vary widely, ranging from those causing no clinical symptoms (for example, balanced translocation), to those of little significance for the affected individual (e.g., Klinefelter syndrome), up to those with a fatal outcome (trisomy 13, 18). As the consequences for the affected families are serious, diagnosis of chromosomal anomalies has always been the main aim of prenatal screening, taking into account all the ethical considerations involved. Amniocentesis was developed over 30 years ago and has been increasingly included in antenatal care for older pregnant women. In Germany, this has had legal consequences. In effect, the gynecologist is obliged to guarantee a chromosomally healthy child and to document the fact that adequate patient information regarding invasive prenatal diagnosis has been provided if the mother is going to be over the age of 34 at the expected date of delivery. In fact, however, only a small percentage of affected fetuses are detected prenatally on the basis of the age indication. In order to offer younger mothers the option of detecting the most frequent chromosomal anomaly—Down syndrome—without the risks of amniocentesis, the triple test was first developed some 10 years ago. The disadvantage of this test is that, at a cut-off level of 1 : 350, it has a false-positive rate of 5–10%, with a detection rate of 60–65%. In recent years, the measurement of nuchal translucency (NT) at 11–13 weeks has been increasingly carried out for early detection of genetic and organ anomalies in younger mothers. The sensitivity of this method for detecting Down syndrome, is 70–80%, with a 5% rate of false-positive findings. In addition to ultrasound scanning, determination of PAPP-A and free β-HCG raises the sensitivity to 90%. This allows many pregnant women above the age of 35 years to avoid invasive prenatal diagnosis, which have their own risks. However, it should be borne in mind here that a sensitivity of 90% means that one in 10 affected fetuses will not be detected. Self-Help Organizations Title: The Genetic Alliance Description: Consortium of support groups dedicated to helping individuals and families affected by genetic disorders. Information and referrals to support groups and genetic services. Scope: National network Telephone: 1–800–336–4363 or 202–966–5557 Fax: 202–966–8553 E-mail: info@geneticalliance.org Title: Chromosome 22 Central Description: Networking and support for parents of children with any chromosome disorder. Supports research. Offers newsletter, literature, phone support and pen pals. Scope: International network Founded: 1996 Address: 2327 Kent Ave., Timmons, Ontario P4N 3 C3, Canada Telephone: 705–268–3099 Fax: 705–268–3099 Web: http://www.c22c.org Definition: This is a structural chromosomal anomaly with a deletion, of varying size, of the distal long arm of chromosome 11. It is associated with multiple congenital malformations and mental retardation. First described in 1973. Etiology: Deletion of the long arm of chromosome 11, as a new mutation or due to balanced translocation in one of the parents. Ultrasound findings: Intrauterine growth restriction, trigonocephaly, microcephaly, dilation of ventricles, holoprosencephaly, hypertelorism, micrognathia, ear anomaly, cardiac and renal anomalies, anomalies of the genitals, clinodactyly, joint contractures. Differential diagnosis: Trisomy 13, trisomy 18, craniostenosis, spina bifida. Prognosis: This depends on the severity of the cardiac anomaly. Survivors suffer moderate to severe mental impairment. Self-Help Organization Title: 11 q Research and Resource Group Description: Mutual support for families of children with structural abnormalities of chromosome 11, including deletions, duplications and translocations. Networking of families, researchers and organizations. Newsletter, conferences, books, videos. Scope: Online Address: Melanie 11 q Research and Resource Group, 6123 A Duncan Rd., Petersberg, VA 23803, United States Web: http://www.11q.net References Fernandez Gonzalez N, Prieto Espunes S, Ibanez Fernandez A, Fernandez Colomer B, Lopez Sastre J, Fernandez Toral J. [Deletion 11 q 23→qter (Jacobsen Syndrome) associated with duodenal atresia and annular pancreas; in Spanish.] An Esp Pediatr 2002; 57: 249–52. Chen CP, Chern SR, Tzen CY, et al. Prenatal diagnosis of de novo distal 11 q deletion associated with sonographic findings of unilateral duplex renal system, pyelectasis and orofacial clefts. Prenat Diagn 2001; 21: 317–20. Chen CP, Liu FF, Jan SW, Chen CP, Lan CC. Partial duplication of 3 q and distal deletion of 11 q in a stillbirth with an omphalocele containing the liver, short limbs, and intrauterine growth retardation. J Med Genet 1996; 33: 615–7. Definition: This is a severe chromosomal disorder, clinically characterized by a severe decrease in muscle tension, severe mental retardation and other anomalies. Mosaic with tetrasomy 12 p. Incidence: Rare condition; only 50 cases have been described. Etiology: Sporadic occurrence. Chromosomal mosaic is difficult to detect in the lymphocytes, but not in fibroblasts and amniotic cells. It correlates with advanced maternal age. Ultrasound findings: Growth restriction, hydramnios, singular umbilical artery and microcephaly are possible findings. Cerebellar hypoplasia, hydrocephalus, craniofacial anomalies such as large forehead, coarse facial features, hypertelorism, ear dysplasia, macrostomia, and elevated palate may also be present. Cardiac anomalies such as VSD, aortic isthmus stenosis, PDA, ASD, and others occur in 25% of cases. Diaphragmatic hernia is seen in 50% of cases, and anal atresia may also be seen. Other anomalies include renal and genital malformations. Extremities may also be involved: shortening of the bones, small, stubby hands and feet, doubling of the big toes. Skin disorders, alopecia and skin pigmentation may also be seen. Differential diagnosis: Wolf-Hirschhorn syndrome, Fryns syndrome, trisomy 9 mosaic, trisomy 18. Clinical management: Karyotyping of various cellular types may be necessary, as in mosaic disorders only 1–3% of lymphocytes from fetal blood may be abnormal. Analyses of amniocytes or placental tissue shows a higher proportion of abnormal cells. Prognosis: 50% die in the prenatal or perinatal stage. The survivors may reach the age of 10–15 years; the oldest patient known is 45 years old, with severe mental retardation. References Chiesa J, Hoffet M, Rousseau O, et al. Pallister-Killian syndrome [i(12p)]: first prenatal diagnosis using cordocentesis in the second trimester confirmed by in situ hybridization. Clin Genet 1998; 54: 294–302. Choo S, Teo SH, Tan M, Yong MH, Ho LY. Tissue-limited mosaicism in Pallister–Killian syndrome: a case in point. J Perinatol 2002; 22: 420–3. Doray B, Girard-Lemaire F, Gasser B, et al. Pallister–Killian syndrome: difficulties of prenatal diagnosis. Prenat Diagn 2002; 22: 470–7. Langford K, Hodgson S, Seller M, Maxwell D. Pallister–Killian syndrome presenting through nuchal translucency screening for trisomy 21. Prenat Diagn 2000; 20: 670–2. Mathieu M, Piussan C, Thepot F, et al. Collaborative study of mosaic tetrasomy 12p or Pallister–Killian syndrome (nineteen fetuses or children). Ann Genet 1997; 40: 45–54. Mowery RP, Stadler MP, Kochmar SJ, McPherson E, Surti U, Hogge WA. The use of interphase FISH for prenatal diagnosis of Pallister–Killian syndrome. Prenat Diagn 1997; 17: 255–65. Paladini D, Borghese A, Arienzo M, Teodoro A, Martinelli P, Nappi C. Prospective ultrasound diagnosis of Pallister–Killian syndrome in the second trimester of pregnancy: the importance of the fetal facial profile. Prenat Diagn 2000; 20): 996–8. Sharland M, Hill L, Patel R, Patton M. Pallister–Killian syndrome diagnosed by chorionic villus sampling. Prenat Diagn 1991; 11: 477–9. Wilson RD, Harrison K, Clarke LA, Yong SL. Tetrasomy 12p (Pallister–Killian syndrome): ultrasound indicators and confirmation by interphase fish. Prenat Diagn 1994; 14: 787–92. Definition: This is the third most frequent fatal chromosomal disorder seen in the prenatal stage. The chromosomes are tripled (instead of the normal doubling), such that the cell nuclei have 69 instead of 46 chromosomes. Incidence: Some 1–2% of all conceptions have triploidy; these cases end mostly as early spontaneous miscarriages. Six percent of fetuses lost as spontaneous abortions up to 28 weeks show triploidy. Birth of a live infant is extremely rare. Sex ratio: M : F = 1.5 : 1. Clinical history: Sporadic occurrence. Elevation of β-HCG and α-fetoprotein (up to four times the median value) is detected. Etiology: An additional complete set of chromosomes leads to a karyotype of 69,XXX or 69,XXY or 69,XYY. Sixty percent of cases are due to fertilization of one ovum with two sperms (paternal origin); 40% arise due to fertilization of a diploid ovum (maternal origin). Teratogens: Not known. Ultrasound findings: Severe intrauterine growth restriction is detected early (beginning at 12–14 weeks). Oligohydramnios, enlarged placenta with cystic regions (partial molar degeneration in triploidy of paternal origin, that of maternal origin usually of normal appearance). CNS: hydrocephalus, holoprosencephaly, neural tube defects, agenesis of the corpus callosum, Arnold-Chiari malformation. Facial features: hypertelorism, cleft palate, cleft lip. Extremities : syndactyly of the third and fourth fingers, club feet. Others: cardiac anomalies, omphalocele, hydronephrosis, genital anomalies. Caution: The placenta may appear normal; the pattern of anomalies is very variable. Differential diagnosis: Trisomy 9, 13, and 18, infections, Neu-Laxova syndrome, Russell-Silver syndrome, Seckel syndrome. Clinical management: Karyotyping and further ultrasound screening, including fetal echocardiography. If the mother decides to continue with the pregnancy, she should be warned of the high risk of developing preeclampsia and hyperthyroidism. Hyperemesis gravidarum and theca lutein cysts of the ovaries are frequent. The β-HCG level is increased, but only in 80% of cases in case of a partial mole. Cesarean section on the basis of a fetal indication should be avoided. Fig. 9.2 Triploidy. Cardiac effusion at 17 + 1 weeks in a fetus with triploidy. Fig. 9.3 Triploidy. Fetal heart showing “single ventricle” in triploidy, at 21 + 3 weeks. Fig. 9.5 Triploidy. Fetal profile at 14 + 5 weeks, showing severe micrognathia in a fetus with triploidy (69,XXX). Fig. 9.6 Triploidy. Fetal profile with severe micrognathia in triploidy at 21 + 3 weeks. Fig. 9.8 Triploidy. Dilation of the cerebellomedullary cistern at 14 + 5 weeks. Fig. 9.9 Triploidy. Tetradactyly in a fetus with triploidy at 14 + 5 weeks. Fig. 9.10 Triploidy. Unilateral multicystic renal dysplasia in a case of triploidy at 17 + 1 weeks. Fig. 9.11 Triploidy. Genitals in a fetus with 69,XXX with hypertrophy of the clitoris at 21 + 3 weeks. Procedure after birth: Intensive medical measures are not indicated. Prognosis: The pregnancy usually ends with an early miscarriage. Otherwise intrauterine demise or death in the early neonatal stage is the general outcome. In case of mosaic, survival into early childhood is rare, with severe mental retardation. Information for the parents: The risk of recurrence is not increased. References Beinder E, Voigt HJ, Jager W, Wildt L. Partial hydatidiform mole in a cytogenetically normal fetus. Geburtshilfe Frauenheilkd 1995; 55: 351–3. Benacerraf BR. Intrauterine growth retardation in the first trimester associated with triploidy. J Ultrasound Med 1988; 7: 153–4. Blaicher W, Ulm B, Ulm MR, Hengstschlager M, Deutinger J, Bernaschek G. Dandy–Walker malformation as sonographic marker for fetal triploidy. Ultraschall Med 2002; 23: 129–33. Harper MA, Ruiz C, Pettenati MJ, Rao PN. Triploid partial molar pregnancy detected through maternal serum alpha-fetoprotein and HCG screening. Obstet Gynecol 1994; 83: 844–6. Jauniaux E. Partial moles: from postnatal to prenatal agnosis. Placenta 1999; 20: 379–88. Jauniaux E, Haider A, Partington C. A case of partial m associated with trisomy 13. Ultrasound Obstet Gynecol 1998; 11: 62–4. Mittal TIC, Vujanic GM, Morrissey BM, Jones A. Triploidy: antenatal sonographic features with postmortem correlation. Prenat Diagn 1998; 18: 1253–62. Pecile V, Demori E, Gambel Benussi D, Dolce S, Amoroso A. Diagnosis of triploidy in metaphases from uncultured amniocytes. Prenat Diagn 2002; 22: 78–9. Philipp T, Kalousek DK. Neural tube defects in missed abortions: embryoscopic and cytogenetic findings. Am J Med Genet 2002; 107: 52–7. Pircon RA, Porto M, Towers CV, Crade M, Gocke SE. Ultrasound findings in pregnancies complicated by fetal triploidy. J Ultrasound Med 1989; 8: 507–11. Ranzini AC, Sharma S, Soriano C, Vintzileos AM. Early diagnosis of triploidy [letter]. Ultrasound Obstet col 1997; 10: 443–4. Rubenstein JB, Swayne LC, Dise CA, Gersen SL, Schwartz JR, Risk A. Placental changes in fetal triploidy syndrome. J Ultrasound Med 1986; 5: 545–50. Wasserman SA, Bofinger MK, Saldana LR. Ultrasound and genetic features of a term triploid pregnancy J Perinatol 1991; 8: 398–401. Yaron Y, Heifetz S, Ochshorn Y, Lehavi O, Orr-Urtreger A. Decreased first trimester PAPP-A is a predictor of adverse pregnancy outcome. Prenat Diagn 2002; 22: 778–82. Definition: Chromosomal disorder with a tripled chromosome 8; postnatal diagnosis is usually a mosaic; patients with pure trisomy 8 are rare. Incidence: As far as chromosomal aberrations are concerned, this is quite a frequent anomaly (> 100 cases have been described); overall, it is a rare disorder. Clinical history/genetics: The occurrence of trisomy 8 and trisomy 8 mosaic is relatively common after chorionic villus sampling. Prognosis of the disorder, only relating to the placenta and that of a real fetal mosaicism is uncertain. Anomalies/ultrasound findings: The clinical manifestations differ considerably; completely normal patients have also been described. Symptoms: overweight, cardiac anomalies, renal and skeletal malformations, often mild to moderate mental retardation. Patients with a low proportion of mosaicism and with normal intelligence have also been described. In addition: arthrogryposis, tetra-amelia, high risk of cancer, in the prenatal stage: thickened nuchal translucency, dilation of the renal calices. Differential diagnosis: Syndromes with arthrogryposis. Prognosis: This is difficult to predict, and depends on the malformations and the proportion of pathological cells (affecting mental development). References Campbell S, Mavrides E, Prefumo F, Presti F, Carvalho JS. Prenatal diagnosis of mosaic trisomy 8 in a fetus with normal nuchal translucency thickness and reversed end-diastolic ductus venosus flow. Ultrasound Obstet Gynecol 2001; 17: 341–3. Danesino C, Pasquali F, Dellavecchia C, Maserati E, Minelli A, Seghezzi L. Constitutional trisomy 8 mosaicism: mechanism of origin, phenotype variability, and risk of malignancies. Am J Med Genet 1998; 80: 540. Gotze A, Krebs P, Stumm M, Wieacker P, Allhoff EP. Trisomy 8 mosaicism in a patient with tetraamelia. Am J Med Genet 1999; 86: 497–8. Jay A, Kilby MD, Roberts E, et al. Prenatal diagnosis of mosaicism for partial trisomy 8: acase report including fetal pathology. Prenat Diagn 1999; 19: 976–9. van Haelst MM, Van Opstal D, Lindhout D, Los FJ. Management of prenatally detected trisomy 8 mosaicism. Prenat Diagn 2001; 21: 1075–8. Definition: An extra triple chromosome 9, frequently as trisomy 9 p. It is associated with multiple anomalies. Usually occurs as mosaic forms, as pure trisomy usually leads to early fetal demise. First described in 1973 by Feingold and Atkins. Ultrasound findings: Growth restriction. Head and face: microcephaly, cerebellar anomaly, ventriculomegaly, Dandy–Walker cysts, neural tube defect, facial clefts, micrognathia, microphthalmia. Extremities: flexion anomaly of the fingers, abnormal fixation of the feet. Thorax and abdomen: cardiac anomaly, liver calcification, singular umbilical artery, renal anomalies, maldescent of the testis. Differential diagnosis: Trisomy 13, trisomy 18, Wolf–Hirschhorn syndrome, triploidy Prognosis: Intrauterine demise or neonatal death is the frequent outcome. Survivors are severely mentally impaired. In mosaic forms, the severity depends on the percentage of affected cells and the organ systems involved. Fig. 9.12 Trisomy 9. Widening of the cerebellomedullary cistern at 16 + 1 weeks (Dandy–Walker variant). Fig. 9.13 Trisomy 9. Echogenic kidneys, with dilation of renal pelvis as well as fetal ascites at 16 + 1 weeks. Hengstschlager M, Bettelheim D, Repa C, Lang S, Deutinger J, Bernaschek G. A fetus with trisomy 9 p and trisomy 10 p originating from unbalanced segregation of a maternal complex chromosome rearrangement t (4;10;9). Fetal Diagn Ther 2002; 17: 243–6. McDuffie RSJ. Complete trisomy 9: case report with ultrasound findings. Am J Perinatol 1994; 11: 80–4. Merino A, De Perdigo A, Nombalais F, Yvinec M, Le Roux MG, Bellec V. Prenatal diagnosis of trisomy 9 mosaicism: two new cases. Prenat Diagn 1993; 13: 1001–7. Murta C, Moron A, Avila M, Franca L, Vargas P. Reverse flow in the umbilical vein in a case of trisomy 9. Ul-trasound Obstet Gynecol 2000; 16: 575–7. Pinette MG, Pan Y, Chard R, Pinette SG, Blackstone J. Prenatal diagnosis of nonmosaic trisomy 9 and related ultrasound findings at 11.7 weeks. J Matern Fetal Med 1998; 7: 48–50. Saura R, Traore W, Taine L, et al. Prenatal diagnosis of trisomy 9: six cases and a review of the literature. Prenat Diagn 1995; 15: 609–14. Tseng JJ, Chou MM, Shih-Chu Ho E. Varix of the portal vein: prenatal diagnosis in a fetus with mosaic trisomy 9 syndrome. Prenat Diagn 2002; 22: 495–7. von Kaisenberg CS, Caliebe A, Krams M, Hackeloer BJ, Jonat W. Absence of 9q22–9qter in trisomy 9 does not prevent a Dandy–Walker phenotype. Am J Med Genet 2000; 95: 425–8. Definition: Additional chromosome 10; usually fatal except when mosaicism is present. Incidence: Seen in about 2% of spontaneous miscarriages. Postnatally, extremely rare. Ultrasound findings: Intrauterine growth restriction, nuchal edema, hypertelorism, cardiac anomaly, cryptorchism. In complete trisomy 10 additionally facial clefts, micrognathia, polydactyly, syndactyly of the toes. Differential diagnosis (of nuchal edema): Trisomy 18, trisomy 21, Turner syndrome, multiple pterygium syndrome, Pena–Shokeir syndrome, Roberts syndrome, Smith–Lemli–Opitz syndrome, and others. Prognosis: Complete trisomy 10 is fatal; infants with mosaic forms may survive with mental impairment and short growth, but life expectancy is severely reduced. References Brizot ML, Schultz R, Patroni LT, Lopes LM, Armbruster-Moraes E, Zugaib M. Trisomy 10: ultrasound features and natural history after first trimester diagnosis. Prenat Diagn 2001; 21: 672–5. Hengstschlager M, Bettelheim D, Repa C, Lang S, Deutinger J, Bernaschek G. A fetus with trisomy 9 p and trisomy 10 p originating from unbalanced segregation of a maternal complex chromosome rearrangement t (4;10;9). Fetal Diagn Ther 2002; 17: 243–6. Knoblauch H, Sommer D, Zimmer C, et al. Fetal trisomy 10 mosaicism: ultrasound, cytogenetic and morphologic findings in early pregnancy. Prenat Diagn 1999; 19: 379–82. Martin-Denavit T, Attia-Sobol J, Theuil J, et al. First prenatal diagnosisofpartial trisomy 10 and partial mon-osomy 15 derived from a maternal translocation (10;15)(q11;q13). Prenat Diagn 2002; 22: 487–8.
9 Chromosomal Disorders
General Considerations
Jacobsen Syndrome (11 q Deletion)
Pallister–Killian Syndrome (Tetrasomy 12 p)
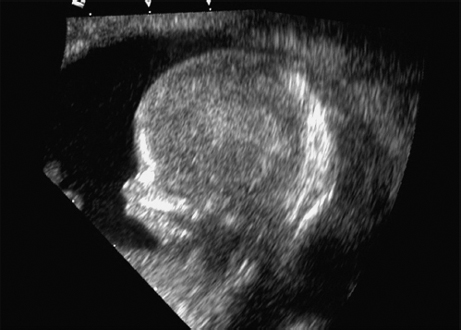
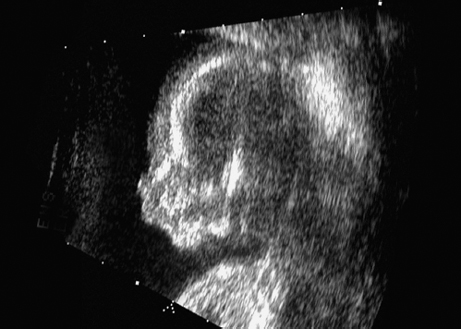
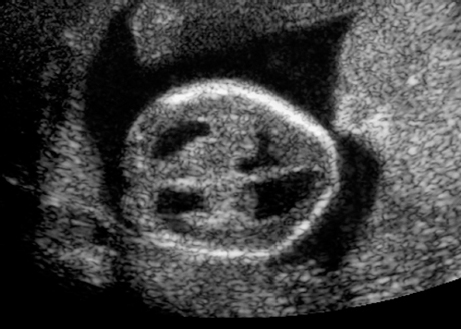
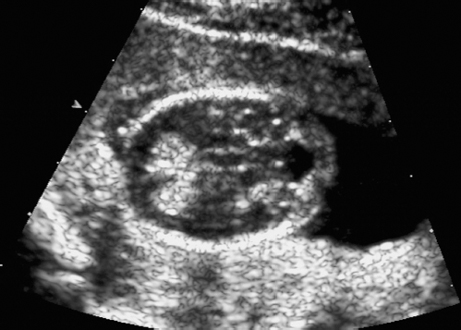
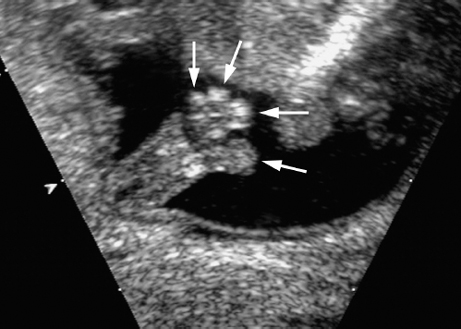
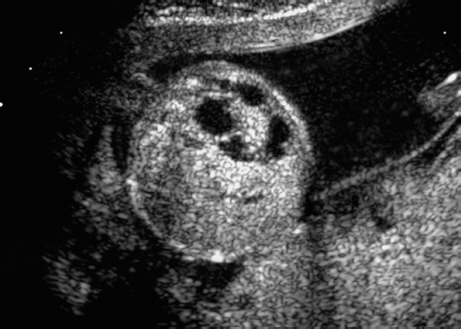
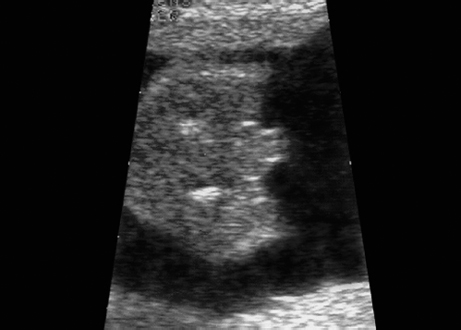
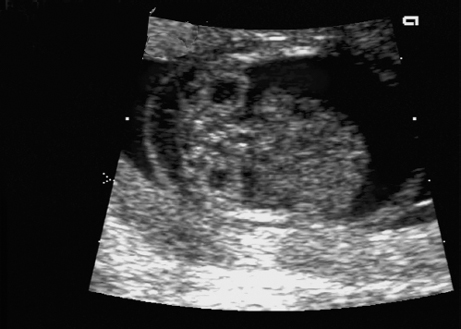
Triploidy
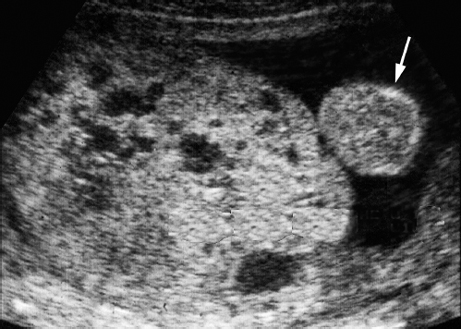
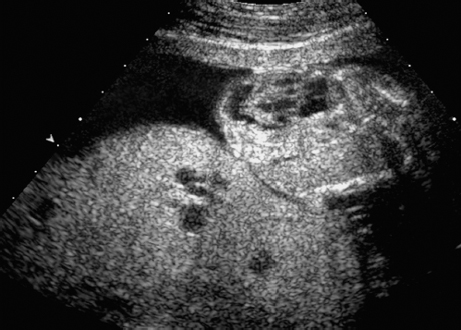
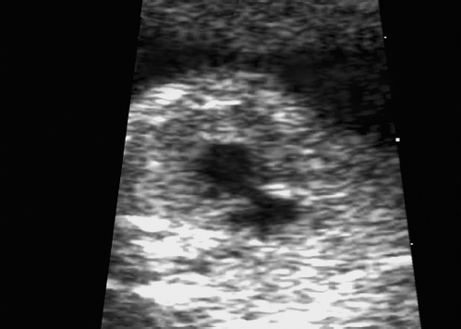
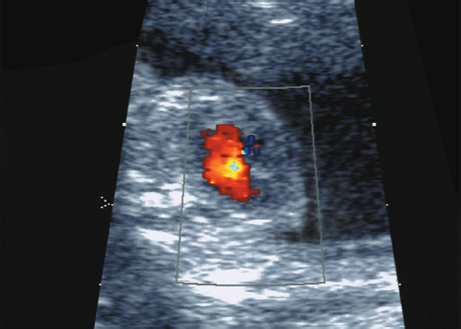
Trisomy 8
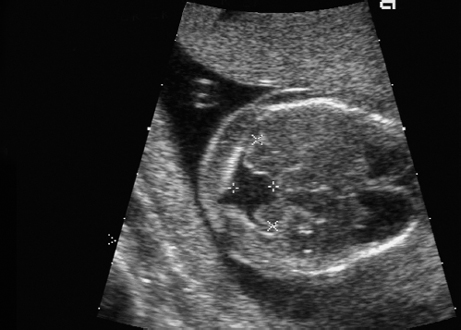
Trisomy 9, Partial Trisomy 9 p
Trisomy 10
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


