Fig. 23.1
Diagram demonstrating histological abnormalities in bone with osteogenesis imperfecta (OI). (a) Normal bone for comparison. (b) Diminished thickness due to impaired periosteal bone formation in OI. Trabeculae are reduced in number and are abnormally thin
Clinical diagnosis is based on increased bone fragility combined with the appropriate signs and symptoms (Table 23.1). A positive family history or the presence of several typical features simplifies diagnosis. However, diagnosis can be challenging in those with only a moderate increase in number of childhood fractures or in those who have gracile bones with thin cortices but no extraskeletal manifestations. Many of these patients have a family history of similar borderline skeletal abnormalities. Analysis of the collagen type 1 genes, either by screening for coding mutations in DNA extracted from the patient’s white blood cells or by analyzing the amount and structure of type 1 procollagen molecules derived from the patient’s cultured fibroblasts, can confirm the diagnosis in around 90 % of cases [7]. However, not all OI genes have been sequenced, and therefore negative chromosomal analysis cannot rule out OI. This has significant social and legal implications in the setting of suspected non-accidental trauma [11, 12] (see Chap. 18).
Table 23.1
Modified Sillence classification of osteogenesis imperfecta
Type | Inheritance | Clinical severity | Typical features | Affected gene(s) |
|---|---|---|---|---|
I | AD | Mild nondeforming | Normal height/mild short stature | COL1A1, COL1A2 |
Normal teeth (except type I) | ||||
Hearing loss | ||||
II | AD, AR | Perinatal lethal | Multiple rib/long bone fractures at birth | COL1A1, COL1A2, CRTAP, LEPRE1, PPIB |
Pronounced deformities | ||||
Lucent skull bones | ||||
Dark sclera | ||||
Very short | ||||
III | AD, AR | Severely and progressively deforming | Triangular face | COL1A1, COL1A2, CRTAP, LEPRE1, PPIB, FKBP10, SERPINH1 |
Severe scoliosis | ||||
Grayish sclera, may normalize | ||||
Dentinogenesis imperfecta | ||||
Moderately short | ||||
Fractures usually by age 2 | ||||
IV | AD, AR | Moderately deforming | Mild to moderate scoliosis | COL1A1, COL1A2, CRTAP, FKBP10, SP7 |
Grayish or white sclera | ||||
Dentinogenesis imperfecta | ||||
V | AD | Moderate to severe bone fragility | Mild to moderate short stature | Unknown |
Mineralized interosseous membrane | ||||
White sclera | ||||
Normal teeth |
Many but not all patients with OI have abnormal sclera, ranging in color from faint gray-blue to deep blue. However, healthy infants can also have dark or bluish sclera, so this finding is not helpful in this age group [7]. Coloration is attributed to scleral thinning or other abnormalities that render visible the vascular layer of the choroid. The cornea may be abnormal as well.
Teeth are abnormal in over one third of patients. They range in color from amber brown to bluish-gray and have an opalescent character. The basic problem lies in the dentin, which is abnormally friable and induces cracking of normal enamel. Permanent teeth are usually less affected than primary teeth. Radiological or histological examination frequently shows abnormalities, even in individuals with apparently normal teeth [13]. Occasionally the disease is completely restricted to the teeth; this condition is termed dentinogenesis imperfecta.
Skin may be thin and translucent; it may bruise easily. The head may be large, with a prominent occipital overhang and eventually a characteristic triangular face. Peripheral joints are hypermobile and lax. Congenital hernias are described in some patients. A defect in homeostasis may precipitate hyperthermia during anesthesia and also cause these children to sweat easily and rarely feel chilled. Conductive or sensorineural deafness occurs occasionally. Arthrogryposis and osteopoikilosis infrequently accompany osteogenesis imperfecta, and several cases of osteogenic sarcoma have been described.
Fractures can occur with the least provocation in some patients and only rarely in others. However, fracture frequency decreases before puberty in severe cases and during and after puberty in all patients. The average number of fractures per individual ranges from less than 1 to 20–30 per year.
Short stature results from several factors. Multiple fractures with resultant bowing may shorten the legs, and repetitive trauma to the growth plate may induce growth plate dysfunction. Most patients also have an intrinsic growth defect.
Classification System
OI was first described in 1788 by a Swedish army surgeon [14]. In 1906, OI was classified into the now obsolete congenita and tarda forms [1, 8]. In 1978, Sillence and coworkers provided the first systematic classification into four clinical types, which were correlated with inheritance patterns: type I (mild OI, blue sclerae, autosomal dominant inheritance), type II (lethal perinatal OI, autosomal recessive inheritance, later subdivided in II-A, -B, and -C based on radiographic features), type III (progressively deforming, sclerae initially blue, autosomal recessive inheritance), and type IV (mild to moderate OI, normal sclerae, autosomal dominant inheritance) [15, 16].
New research unraveling the genetic and clinical complexity of OI has made additional classification possible, but the Sillence scheme is still clinically useful [17]. After the discovery of heterozygous collagen type I gene mutations in all types of OI [18, 19], the Sillence criteria were used mainly to describe the severity of disease and differences in clinical expression, rather than the inheritance pattern. With the subsequent discovery that some patients with lethal or severe OI have normal type I collagen [20], the Sillence classification was expanded, first to include seven genetic types [7] and then to include eight [21]. Type V was added because of its characteristic clinical and radiological features, whereas the histology of type VI is unique. Types VII and VIII are genetically distinct, both with autosomal recessive inheritance and mutations in, respectively, the CRTAP and LEPRE1 genes [17]. The Nosology Group of the International Skeletal Dysplasia Society has retained the Sillence classification as the prototypic and universally accepted way to classify OI and avoids direct molecular reference (see Table 23.1).
The relatively mild types I and IV comprise considerably more than half of patients with OI. It is difficult to predict exactly what form this disease will take. About 20 % of patients with mild OI (especially those with type IV) have fractures at birth. Type III accounts for around 15 % of patients with OI, while type V accounts for between 4 and 5 % of all cases. The incidence of type II is reported at 1–2 per 100,000 [7, 8].
Management of OI is multidisciplinary. Subspecialty management of hearing, dental, and pulmonary abnormalities may be necessary. Skeletal abnormalities are addressed with medications such as bisphosphonates and recombinant human growth hormone, as well as surgical procedures and physical rehabilitation. Although some form of bisphosphonate therapy constitutes standard treatment, the specific drug and duration of treatment are subject to debate. Furthermore, it is difficult to predict which patients will respond with increased bone density and decreased incidence of fracture. Promising cellular therapies and new drug regimens are being developed.
Orthopedic treatment includes casting, rod placement, and osteotomies (cutting and realigning the bone); choice of procedure depends on the fracture type, disease severity, and patient’s functional status. If routine casting is unsuccessful, intramedullary rods may be placed. Fragmentation may be required for difficult cases and is usually successful. This involves cutting a bowed and deformed bone into segments and then realigning them on a rigid or expandable rod. Complications following this and other procedures include rod breakage, uncoupling, extrusion, interference with growth plate function, and fracture at the end of a metallic device. Nonunion occasionally occurs, but in general fractures and osteotomies heal well. Management of scoliosis is complicated by extreme bone fragility.
Imaging
An excessive number of Wormian, or intrasutural, bones (exceeding the upper normal of eight to ten) are a typical feature in the skull [7, 22] (Fig. 23.2). Bones may be diffusely osteoporotic, with healing or healed fractures. Joint abnormalities such as genu valgum and coxa vara can result from both laxity and fracture deformity, while loss of structural strength can lead to acetabular protrusion (see Chap. 3).
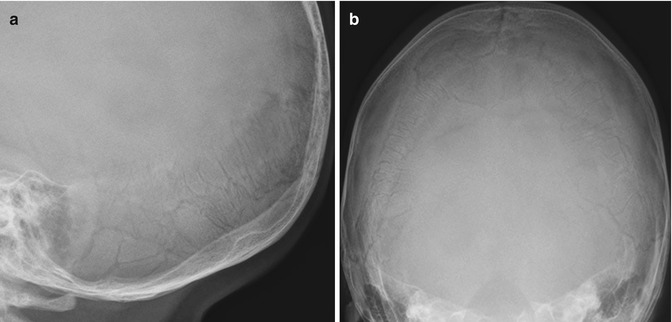
Fig. 23.2
(a, b) Multiple Wormian bones in the lambdoid suture in a 17-month-old boy with OI type I
Although there is considerable overlap, some imaging manifestations are more specific to the type of OI and are discussed below (Box 23.1).
Box 23.1: Osteogenesis Imperfecta: Radiographic Features
All | Excessive number of Wormian bones (>8–10) |
Diffuse osteoporosis | |
I | Mild nondeforming |
Slender ribs/long bones, no major bone deformity, normal or mild short stature | |
Fractures after 2–3 years old (may be preceded by bowing) | |
Diaphyseal, transverse, nondisplaced; often lower extremity | |
Decreased frequency after puberty | |
Vertebral fractures common | |
II | Perinatal lethal |
Beaded ribs, crumpled femurs, severely short stature | |
Short, broad long bones | |
Severe biconcave vertebrae | |
III | Severely and progressively deforming |
Gracile, mildly short long bones, very short stature | |
Angulation at metadiaphysis | |
Codfish vertebrae, progressive severe kyphoscoliosis | |
Popcorn metaphyseal calcifications in up to 50 % | |
Usually fractures at birth | |
IV | Moderately deforming |
Mild to moderate bone fragility, variable short stature | |
Up to ¼ have fractures at birth | |
Radiographs resemble type I but may have popcorn calcifications in metaphyses | |
V | Moderate to severe bone fragility |
Hyperplastic callus resembles osteosarcoma | |
Variable short stature | |
Calcification of interosseous membrane at the forearm, causing dislocation of the radial head |
OI Type I (Mild Nondeforming)
This is the mildest and most common form of OI (see Table 23.1). It is associated with blue sclerae that persist throughout life, hearing loss in half of patients by 40 years of age, joint hypermobility, postfracture deformity, and easy bruising. Fracture incidence and severity vary. They often occur only after initiation of weight bearing, around 2 or 3 years of age. Children with OI who are younger than 5 years of age usually have diaphyseal, transverse, nondisplaced, and often lower extremity fractures, whereas normal children of the same age tend to fracture in the dia/metaphyses of the upper extremities [23]. Fractures continue throughout childhood but decrease in frequency after puberty. The “A” subtype is associated with dentin abnormality.
Radiographs usually demonstrate osteoporosis and slender ribs and long bones (Fig. 23.3). Mild anterolateral bowing can precede fractures (Fig. 23.4). Vertebral fractures are common and can lead to mild scoliosis (see Chap. 3) (Fig. 23.5). As with other forms of OI, Wormian (or intrasutural) bones are common (see Fig. 23.2).
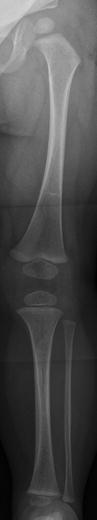
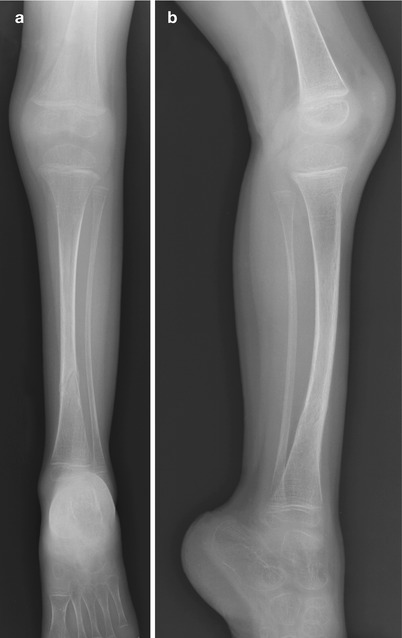
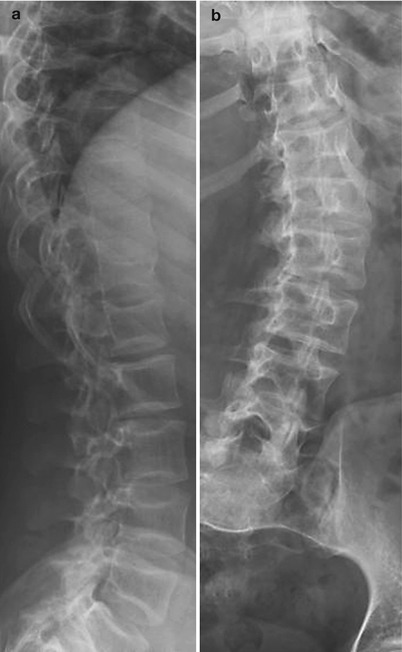
Fig. 23.3
Osteoporosis and slender long bones in a 2-year-old girl with OI type I
Fig. 23.4
OI type I in a 3-year-old. (a, b) The bones are gracile and show mild tibial bowing. Healing distal tibial fracture
Fig. 23.5
OI type I in a 12-year-old girl. (a) Osteoporosis and multiple vertebral fractures. (b) Mild scoliosis
OI Type II (Perinatal Lethal)
This is characterized by blue sclerae and represents the most common second trimester ultrasound diagnosis of short, bent limbs [1]. Healing intrauterine fractures result in a beaded appearance of the ribs and crumpled femurs; deformity is obvious at birth (Fig. 23.6). Ribs are small and dysplastic due to multiple fractures, and this may result in a hypoplastic thorax with reduced lung volume and eventual respiratory failure and death. The bones of the extremities are short and wide, with evidence of periosteal new bone formation, callus, and healing and unhealed fractures. The increased bone width is largely due to remodeling after fracture healing, resulting in a blocklike, rectangular shaft. Impacted and torus fractures lead to wrinkled, crumpled, and thick cortices. Femoral and tibial bowing is typical. Severe biconcave compression deformity of the vertebral bodies may become so severe that the flattened, diminutive, osteoporotic vertebrae can barely be recognized between the intervertebral discs.
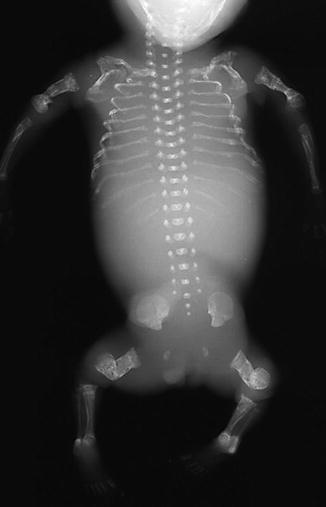
Fig. 23.6
OI type II in a fetus, whose gestation was terminated at 24 weeks. Multiple old and recent fractures of the extremities and ribs. Squaring of long bones due to periosteal callus in response to intrauterine fractures
Radiographs of the most severe type A subgroup demonstrate absent ossification of the skull vault (Fig. 23.7), marked osteoporosis, broad ribs from multiple fractures, short and crumpled femurs with no metadiaphyseal differentiation, vertebral collapse, and angulated tibias [24]. The type B subgroup, which must be differentiated from type III in the perinatal period, has beaded ribs from discrete healing fractures, along with angulated tibias, but there is recognizable distinction between the metadiaphysis and the diaphysis. The type C subgroup is rare. It too lacks skull vault ossification but has unusually dense stippling throughout the skeleton, possibly secondary to healing fractures.
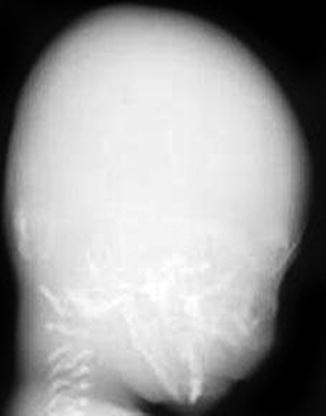
Fig. 23.7
Defective calvarial ossification in OI type IIA
OI Type III (Severely and Progressively Deforming)
This is the most severe form in children that survive the neonatal period. The bones are extremely slender at birth, often with fractures and poor growth. By 2 years of age, numerous fractures are evident. Blue sclerae at birth and infancy later normalize. Hearing loss occurs early, teeth are abnormal, and the face is triangular with frontal bossing.
Radiographs demonstrate severe osteoporosis, poor ossification of the skull with wide sutures, multiple Wormian bones, and large fontanelles (Fig. 23.8). The long bones are gracile and mildly shortened, with marked angulation and subsequent widening of the meta- and diaphyses (Figs. 23.9 and 23.10). The vertebral bodies develop a characteristic codfish shape, and there is progressive kyphoscoliosis.
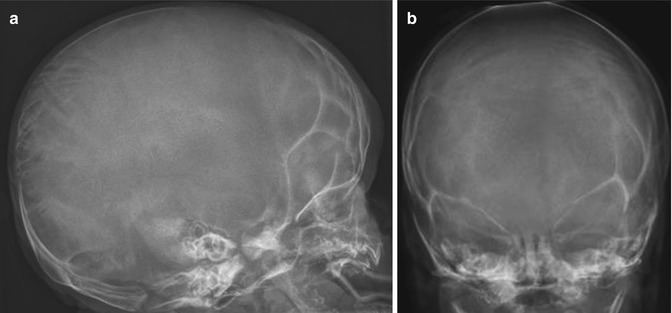
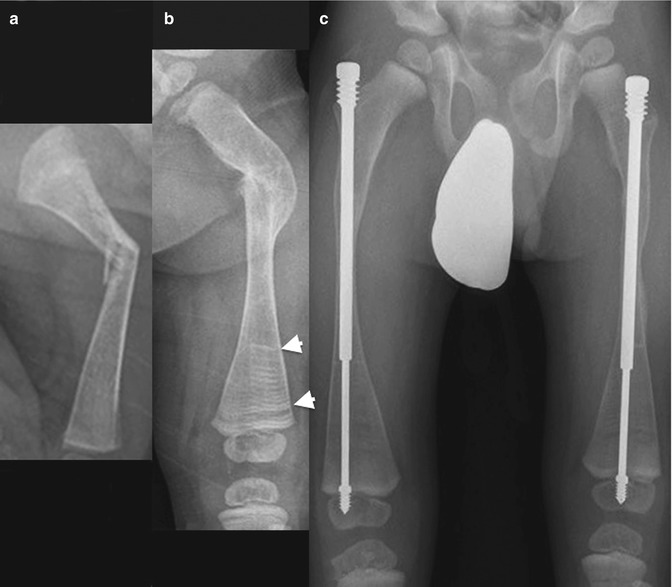
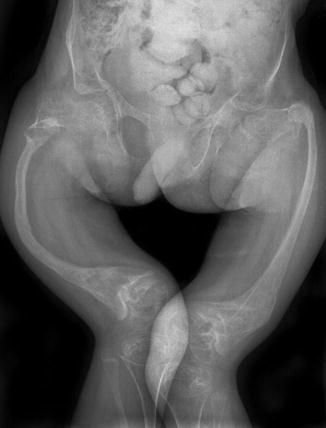
Fig. 23.8
OI type III in a 3-week-old boy. (a, b) Under-mineralized skull with wide sutures and multiple Wormian bones
Fig. 23.9
Progressive changes in the femur of a boy with OI type III. (a) Recent femoral fracture when 3 weeks old. (b) At age 1.5 years, the fracture has healed with deformity. (c) At age 4 years, both femurs have been pinned, and bones are osteoporotic. Horizontal metaphyseal lines (arrowheads) due to bisphosphonate treatment
Fig. 23.10
Marked osteoporosis and deformity in a 9-year-old boy with OI type III, who was born with multiple fractures. The femurs are gracile, curved, and irregular due to numerous incomplete fractures
“Popcorn” metaphyseal calcifications are encountered in up to one half of older children with type III OI and are occasionally seen in type IV. Resembling accumulations of popcorn, these lesions consist of multiple discrete conglomerations of small cystic lucencies, demarcated by sclerotic borders and sometimes containing central calcifications (Fig. 23.11) [25]. They are characteristically located in the lower extremities, especially at the knees, and can cause severe growth dysfunction. The metaphysis is typically expanded, and the growth plates partially or completely disrupted, with popcorn nodules extending into the epiphysis. Popcorn calcifications are not present at birth but develop as the patient grows, remodeling and disappearing with maturity. Their location and evolution suggest they may result from trauma to abnormal bone.
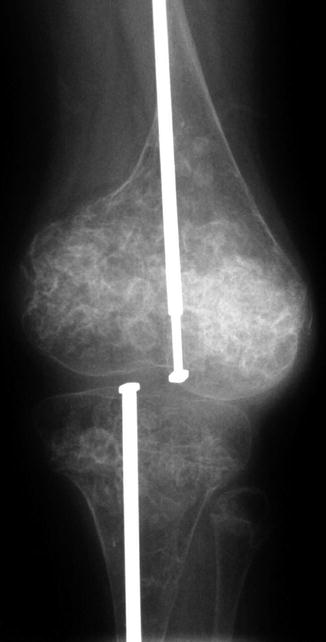
Fig. 23.11
Metaphyseal “popcorn” calcifications in a 12-year-old boy with OI (Courtesy of Sandra L. Wootton-Gorges)
OI Type IV (Moderately Deforming)
This represents a heterogeneous group characterized by mild to moderate osseous fragility and mild to moderate bone deformities. Sclerae are usually grayish or white (although they can be blue in infancy and childhood), and hearing is normal. Dental problems, osteoporosis, and fracture incidence vary, and the initial fractures occur at birth in up to one fourth of patients. Radiographs of these patients resemble those with type I OI (Fig. 23.12). However, popcorn calcifications are encountered in about 10 % of patients with type IV OI.
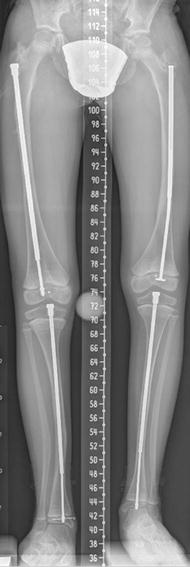
Fig. 23.12
Multiple healed fractures in an 11-year-old boy with OI type IV and resultant mild leg length discrepancy. Bones are severely osteopenic, with thin cortices and trabeculae. Impaired periosteal growth has led to thin diaphyses with relatively normal metaphyses
OI Type V (Moderate to Severe Bone Fragility)
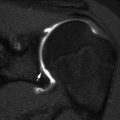
This form is characterized by calcification of interosseous membranes and/or hypertrophic callus [26]. Patients may be somewhat short but have normal teeth and sclerae. There is no evidence of a collagen type 1 abnormality [17]. The interosseous membrane at the forearm calcifies early in life, limiting hand movements and eventually causing the radial head to dislocate. After fractures or surgical procedures, hyperplastic callus can mimic osteosarcoma [27] (Fig. 23.13).
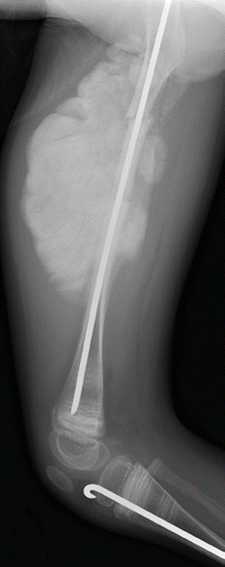
Fig. 23.13
Hyperplastic callus formation in a 13-year-old boy with OI type V and an upper femoral fracture
Differential Diagnosis
Several primary skeletal disorders can be confused with OI (Table 23.2), although chromosomal analysis will often differentiate between them. Radiographs of patients with Bruck syndrome resemble OI, but elbow contractures are present at birth and cubital webs develop [24]. Osteoporosis-pseudoglioma syndrome is similar to OI and was previously called ocular OI. Panostotic fibrous dysplasia involves all bones and is the extreme form of polyostotic fibrous dysplasia. Idiopathic hyperphosphatasia (juvenile Paget disease) is characterized by severe bone fragility and strikingly increased bone turnover [7], along with very high alkaline phosphatase activity. The presentation of hypophosphatasia varies widely, from a severe perinatally lethal form with absent mineralization to pathological fractures that develop only in late childhood [28]. Cole-Carpenter syndrome is characterized by severe bone fragility and is distinguished from OI by the presence of craniosynostosis. In idiopathic juvenile osteoporosis, osteoporosis leads to fractures and collapse of vertebral bodies around age 5, with spontaneous improvement around puberty.
Table 23.2
Skeletal disorders that resemble osteogenesis imperfecta
Disorder | Inheritance | Severity of bone fragility/deformity | Characteristics | Genetic defect |
|---|---|---|---|---|
Bruck syndrome | AR | Moderate to severe | Congenital joint contractures | Telopeptide lysyl hydroxylase deficiency |
Osteoporosis-pseudoglioma syndrome | AR | Moderate | Congenital blindness | LRP5 |
Panostotic fibrous dysplasia | None (somatic mutation) | Severe | Cystic or ground-glass lesions in all bones | GNAS |
Idiopathic hyperphosphatasia | AR | Severe | Raised alkaline phosphatase activity | TNFRSF11B |
Wide diaphyses | ||||
Thick calvarium | ||||
Hypophosphatasia | AR, AD | Mild to severe | Low alkaline phosphatase activity | ALPL |
Cole-Carpenter syndrome | Unknown | Severe | Craniosynostosis Ocular proptosis | Unknown |
Idiopathic juvenile osteoporosis | None | Mild to moderate | No extraskeletal abnormalities | Unknown |
Non-accidental trauma can produce a variety of injuries and injury patterns in children, but none are completely pathognomonic [29]. Fractures are observed in approximately 30 % of abused children and are even more common in infants [29] (see Chap. 18). Collagen type 1 analysis can be useful if positive, but a negative test is by no means evidence for child abuse. Differentiating mild OI from non-accidental trauma is challenging if genetic analysis is normal, there is no family history, and teeth and sclerae are normal. Measurement of bone-mineral density with dual-energy X-ray absorptiometry (DXA) has been proposed to help in the differential diagnosis, but technical issues, difficulties with interpretation, and lack of normative data limit its use in children (see Chap. 26) [30].
2 Neurofibromatosis
Neurofibromatosis is a multisystem disorder with both tumor and non-tumor manifestations. Although anecdotal descriptions of neurofibromatosis date back centuries, von Recklinghausen and Wishart were the first to categorize neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2) [31]. Based on genetic and molecular studies, three distinct forms have been recognized: NF1, NF2, and schwannomatosis.
NF1, previously known as von Recklinghausen or peripheral neurofibromatosis, is most common, with an estimated worldwide prevalence of 1 in 3,500 [32, 33]. An autosomal dominant disorder, it is caused by mutations in the NF1 gene that encodes the tumor suppressor protein neurofibromin [34–36]. This gene is located on chromosome 17 and has 100 % penetrance [36]. Around 50 % of patients with NF1 have inherited the mutation, while the rest have a de novo NF1 mutation. The disorder affects neuroectoderm, mesoderm, and possibly also endocrine organ systems, resulting in abnormalities of skin, nervous system, bones/cartilage, and select solid organs. Musculoskeletal abnormalities are found in about 50 % of patients and principally result from mesodermal dysplasia. Neurofibromas are also very common.
Neurofibromatosis type 2 (bilateral acoustic neurofibromatosis, central neurofibromatosis) is an autosomal dominant inherited tumor predisposition syndrome that occurs in around 1 in 40–60,000. It is caused by mutations in the NF2 gene on chromosome 22. Bilateral vestibular schwannomas are the hallmark NF2 lesions and are seen in nearly all affected individuals by age 30.
Schwannomatosis is a rare disorder that may be associated with the SMARCB1 gene on chromosome 22 (different from the NF2 gene), with an estimated prevalence of about 1 in 40,000 [37]. Most cases are thought to represent new mutations, and only 15 % are familial. In contrast to NF1 and NF2, penetrance is incomplete [31]. With schwannomatosis, patients develop multiple schwannomas everywhere except the vestibular nerve. Neurofibromas are absent.
Hallmarks of neurofibromatosis type 1 are café-au-lait macules and multiple benign cutaneous neurofibromas, but the disorder can affect nearly every organ system. Clinical manifestations are highly variable between unrelated individuals and within the same family and are classified as tumor or non-tumor [31].
Non-tumor manifestations of NF1 commonly affect the skin, central nervous system, vessels, and bones. Café-au-lait macules are hyperpigmented, well-circumscribed lesions that can be present at birth but usually increase in number during the first 2 years of life and then increase in size as the child grows. Although 15 % of the general population has one or two café-au-lait pigmentations, very few people have more than two. Skinfold freckling, which is seen in most patients by age 10, occurs at sites of skin friction, such as the axilla, groin, and neck. High-signal-intensity lesions on T2-weighted (T2-W) images, referred to as (unidentified) bright objects, are frequently observed, typically in the basal ganglia and cerebellar white matter. Most are well demarcated and non-enhancing and diminish with age. They may show mass effect. Learning disabilities are fairly common, and seizures occur in around 5 % of patients. Additional central nervous system findings include aqueductal stenosis, Chiari type 1 malformation, and macrocephaly. Glaucoma, congenital heart disease, hypertension secondary to renal arterial dysplasia, and cerebrovascular disease (stenosis or aneurysms) are also encountered.
Neurofibromas can infiltrate the bladder, gastrointestinal tract, or retroperitoneum. The incidence of pheochromocytoma is increased, and pulmonary fibrosis as well as pulmonic stenosis and other congenital heart lesions have been reported. Skeletal findings include scoliosis, short stature, pseudarthrosis of long bones, sphenoid wing dysplasia, focal osseous lesions, pseudarthrosis, and osteopenia.
The most common tumor in NF1 is the neurofibroma (Box 23.2). This benign hamartomatous tumor is composed of Schwann cells, perineural cells, mast cells, and fibroblasts. The lesions are classified as cutaneous or plexiform, with marked differences in growth patterns and size. Cutaneous neurofibromas are small masses often associated with a single nerve ending. The localized neurofibroma is the most common form of neurofibroma encountered in patients with NF1, but most localized neurofibromas occur in patients who do not have NF1. When associated with a larger nerve, a neurofibroma may infiltrate the dermis and appear as a diffuse mass. Plexiform neurofibromas (Fig. 23.14) are much larger tumors and are usually associated with major nerve trunks and plexi. About one third of patients with NF1 develop clinically detectable plexiform neurofibromas [36]. These bulky masses may cause disfigurement, erode adjacent bones, or cause respiratory or gastrointestinal obstruction. Locally invasive, they are composed of an acid mucopolysaccharide myxomatous matrix that infiltrates normal axonal structures. They can become very large and have a marked potential for malignant transformation. The lifetime risk of malignant degeneration of a peripheral plexiform neurofibroma is estimated at 10–13 % [38] (see Chap. 22).
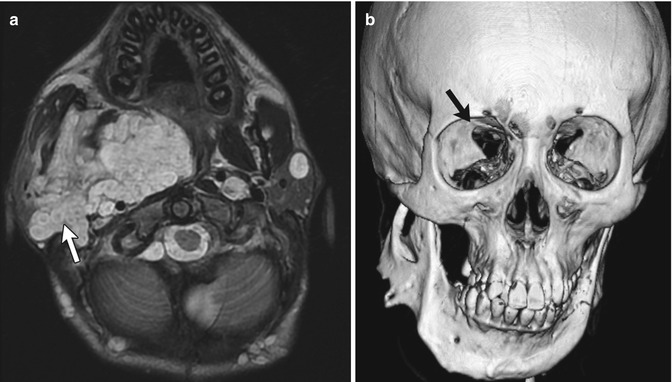
Fig. 23.14
Plexiform neurofibroma and sphenoid wing dysplasia in a 16-year-old male with neurofibromatosis type 1 (NF1). (a) Axial T2-W image shows plexiform neurofibroma involving infratemporal, pterygopalatine, and masticator spaces. Note multiple target signs (white arrow). There are multiple additional smaller neurofibromas in the subcutaneous tissues and left parotid gland. (b) 3D-CT shows mandibular remodeling from the plexiform neurofibroma, and also sphenoid wing dysplasia (black arrow)
Box 23.2: Imaging Features of Neurofibromas
Localized | Well-circumscribed, fusiform mass continuous with nerve |
Diffuse | Infiltrative or plaquelike, elevating skin/thickening dermis |
Plexiform | Ropelike mass along major nerve trunks |
Likely to be benign if | |
Target sign on T2 | Hypointense center, hyperintense periphery |
Reverse target sign on enhanced T1 | Enhancing center, hypointense periphery |
Other tumors encountered in NF1 include Lisch nodules, optic gliomas, and various other malignancies. Lisch nodules (hamartomas that affect the iris) are specific to NF1. Fifteen percent of patients with NF1 develop optic pathway gliomas, but only around 5 % are symptomatic, usually during childhood. Additionally, patients with NF1 have an increased incidence of malignancies such as Wilms tumor, liposarcoma, medullary carcinoma of the thyroid, and hepatocellular carcinoma.
The National Institutes of Health (NIH) has established diagnostic criteria for NF1. These require at least two of the following for diagnosis:
1.
Six or more café-au-lait macules (>5 mm in children and >15 mm in adults)
2.
Two or more neurofibromas of any type or one plexiform neurofibroma
3.
Axillary or groin freckling
4.
Optic pathway glioma
5.
Two or more Lisch nodules
6.
Bony dysplasia
Comprehensive NF1 mutation screening detects mutation in more than 92 % of patients fulfilling the criteria outlined above. Molecular testing for NF1 can be helpful in equivocal cases, particularly in very young children with a negative family history and in whom the clinical criteria are incomplete. Molecular testing is also useful for adults with a mild phenotype, such as café-au-lait macules and axillary freckles but no neurofibromas.
Management of patients with NF1 includes active surveillance for non-tumor and tumor manifestations of the disease. Surgery is reserved for tumors that cause severe symptoms and for those that become malignant. Complete resection of plexiform neurofibromas is usually not possible, and recurrence is common.
Neurofibromatosis type 2 is characterized by schwannomas, which typically affect both vestibular nerves and are associated with symptoms of hearing loss, tinnitus, and impaired balance [39]. The average age of onset is 18–24 years, but about 15 % of patients present earlier with a wide variety of complications. These include orbital meningiomas, retinal hamartomas, and visual loss due to cataracts; seizures due to intracranial meningiomas; and myelopathy due to spinal tumors or cutaneous schwannomas. Childhood mononeuropathy frequently presents as a persistent facial palsy, a squint (third nerve palsy) or hand/foot drop [31].
Manchester diagnostic criteria for NF2 [40] include:
1.
Bilateral vestibular schwannoma
(a)
First-degree relative with NF2 and unilateral vestibular schwannoma or any two of the following: meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities
Or
(b)
Unilateral vestibular schwannoma and any two of the following: meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities
Or
(c)
Multiple meningiomas (at least two) and unilateral vestibular schwannoma or any two of the following: glioma, neurofibroma, schwannoma, cataract
2.1 Imaging (Box 23.3)
Box 23.3: Radiographic Features of NF1
Dystrophic scoliosis | 4–6 vertebral levels |
Kyphosis | |
Neural foraminal widening, scalloped vertebrae | |
Spindled transverse processes, penciled ribs | |
Concave vertebral scalloping | |
Paravertebral mass: neurofibroma, meningeal ectasia, lateral meningocele | |
Ribs | Penciled or ribbonlike |
Fractures | Narrowed or cystic/hourglass expansion of medullary canal |
Sclerotic, tapered ends of bone | |
Segmental sclerosis at fracture site | |
Anterolateral bowing, pseudarthrosis of tibia | |
Numerous nonossifying fibromas/fibrous cortical defects | |
Neurofibromas
These are best imaged with magnetic resonance imaging (MRI). Cutaneous neurofibromas may be localized or diffuse. Localized neurofibromas are well-circumscribed, elongated, or fusiform masses that are continuous with the affected nerve. They usually demonstrate nonspecific signal intensity and variable enhancement. The classic target sign (see Fig. 23.14), which is almost pathognomonic, is evident on T2-W images: hyperintense myxoid peripheral material surrounds a relatively hypointense fibrous center. The “reverse target” sign may be seen on T1-W gadolinium-enhanced images and demonstrates an enhancing center and relatively little peripheral enhancement [41]. Diffuse neurofibromas (Fig. 23.15) grow in either an infiltrative/reticulated branching pattern or as a plaquelike area that elevates the skin and thickens the underlying dermis. These lesions enhance.
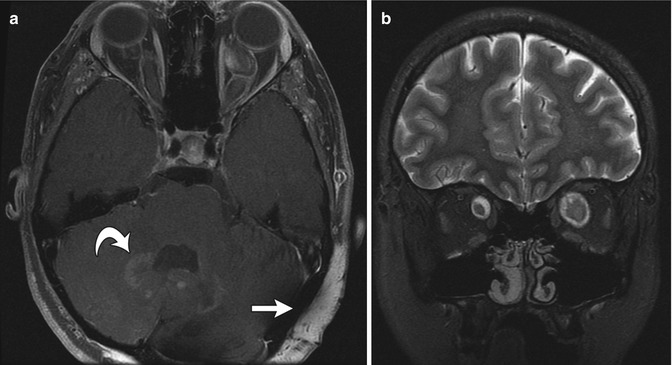
Fig. 23.15
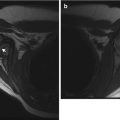
Diffuse neurofibroma and bilateral optic gliomas in a 16-year-old with NF1. (a) Axial T1-W FS GE image shows optic glioma on the left and a diffuse, enhancing, neurofibroma (straight arrow). There are also NF spots (curved arrow) in the cerebellar white matter. (b) Coronal T2-W FS image shows optic nerve gliomas are bilateral, left larger than right (Courtesy of Sandra L. Wootton-Gorges)
Plexiform neurofibromas are bulky, ropelike masses that extend along the sympathetic chain, vagus and phrenic nerves, and major nerve trunks of the extremities. Radiographs play little role in imaging, except in the chest, where they appear as mediastinal or paravertebral soft tissue masses. On MRI, they are best appreciated on fat-suppressed (FS) T2-W sequences, where they appear as multinodular, confluent masses, often with multiple target signs (see Chap. 22 for additional discussion of these tumors). Plexiform neurofibromas are almost pathognomonic for NF1.
Spine
The spine is abnormal in up to 50 % of patients with NF1, and of those with spinal involvement, the thoracic segment is affected in 40 % and the cervical in 30 % [42, 43]. Scoliosis is common, occurring in about 10–69 % of patients. Cervical or upper thoracic kyphosis is another feature of NF1. Vertebral bodies may demonstrate scalloping, and individual vertebrae may demonstrate abnormal pedicles and transverse processes.
Two forms of scoliosis are found in NF1: nondystrophic and dystrophic. Nondystrophic scoliosis is usually radiographically indistinguishable from the idiopathic condition [44], except that the curve typically affects younger patients and may progress to the dystrophic form. Dystrophic scoliosis is often accompanied with neuroforaminal widening, scalloped vertebrae, spindled transverse processes, and penciled ribs; it typically affects only four to six vertebrae and is often associated with kyphosis (which may be more pronounced than the scoliosis) (Figs. 23.16 and 23.17). Dystrophic scoliosis is a hallmark of NF1 and has a poor prognosis. Over 40 % of those with scoliosis also have cervical spine abnormalities. The severity of the curve is unrelated to the degree of skin pigmentation or the presence of subcutaneous tumors.
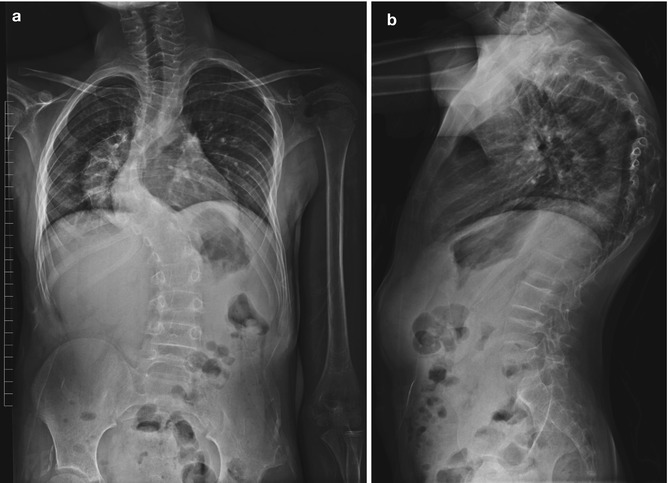
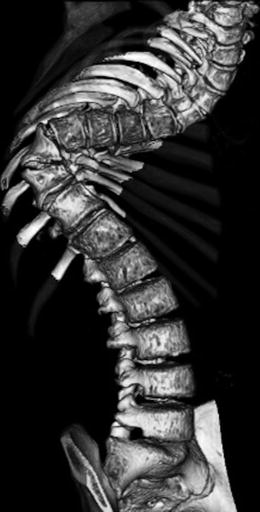
Fig. 23.16
Dystrophic kyphoscoliosis in a teenager with NF1. Short-segment thoracic curvature is associated with narrow posterior ribs (Courtesy of Rebecca Stein-Wexler)
Fig. 23.17
Dystrophic kyphoscoliosis in a patient with NF1. 3D-reformatted CT shows short-segment kyphoscoliosis with dysmorphic vertebrae at the apex of the curve (Courtesy of Sandra L Wootton-Gorges)
The kyphosis associated with neurofibromatosis is often severe but is unusually flexible when traction is applied. In the cervical spine, the anterior aspects of the vertebrae at the apex of the kyphosis are often sharp and pointed, surrounded by hamartomatous tissue.
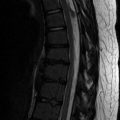
Concave scalloping of the vertebral body may occur on the anterior, posterior, or lateral surfaces. Dural ectasia and transmitted pulsations are thought to cause most posterior vertebral body scalloping (Fig. 23.18), although if only one or two levels are involved, an intradural mass may be responsible. The scalloped, dysplastic vertebral body of NF1 must be differentiated from that caused by intraspinal cysts or neoplasms, hydrosyringomyelia, hydrocephalus, achondroplasia, Melnick-Needles syndrome, and the dural ectasia of the Marfan and Ehlers-Danlos syndromes.
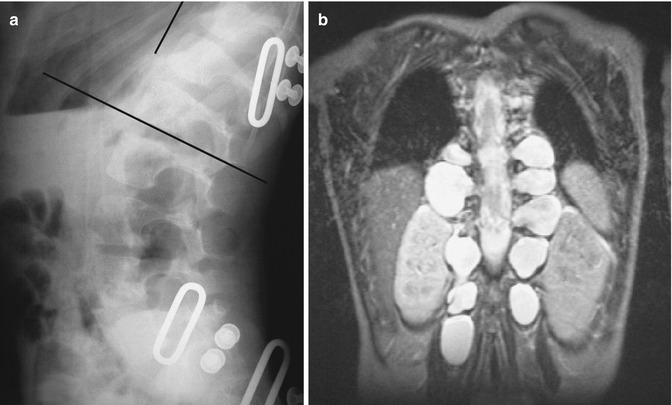
Fig. 23.18
Lateral meningoceles in a patient with NF1. (a) Lateral radiograph demonstrates enlargement of multiple neural foramina along with posterior scalloping of multiple vertebral bodies. (b) Coronal T2-W MRI shows fluid-filled sacs protruding laterally from every neural foramen (Courtesy of Rebecca Stein-Wexler)
Agenesis or hypoplasia of the vertebral pedicles has been described in NF1 but also occurs in otherwise normal spines. The anomaly may be unilateral or bilateral and may involve multiple levels. Pedicle thinning, elongation, sclerosis, and hypoplasia result from mesodermal dysplasia, and the transverse processes are often abnormal. Generalized trabecular coarsening is common, but spondylolisthesis is rare.
Paravertebral masses generally represent either neurofibromas or lateral meningeal ectasia. If the latter enlarge, they may become lateral meningoceles. Foraminal widening or pediculate erosion is seen in 13 % of patients with NF1, usually secondary to primary dysplastic underdevelopment of the pedicles, dumbbell neurofibromas (Fig. 23.19), or lateral meningeal ectasia/meningoceles (see Fig. 23.18). Increased intraspinal volume, hydrosyringomyelia, dural ectasia, and intraspinal neoplasm may alternatively be present. Paraplegia in children usually results from spinal deformity, whereas in adults it tends to result from a tumor.
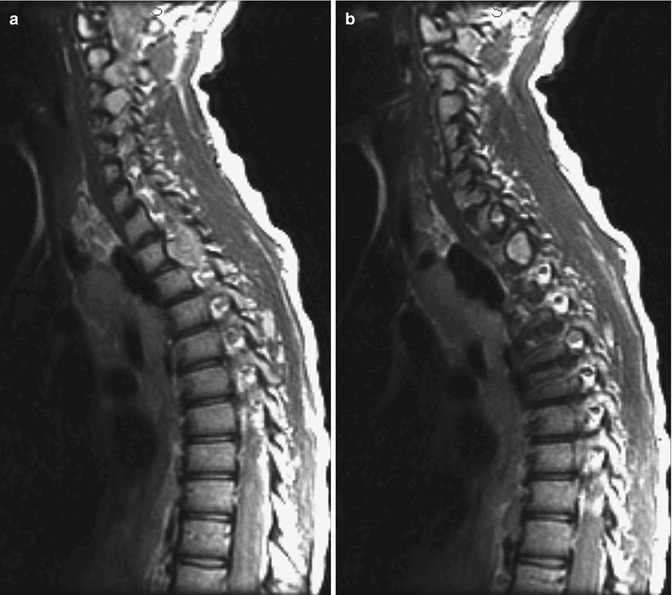
Fig. 23.19
Dumbbell neurofibroma in a patient with NF1. (a, b) Sagittal T2-W images show focal posterior scalloping secondary to a neurofibroma with both intraspinal and neural foraminal components (Courtesy of Rebecca Stein-Wexler)
Computed tomography (CT), especially with its potential for three-dimensional (3D) reconstructions, allows exquisite delineation of dystrophic kyphoscoliosis (see Fig. 23.17). Spine MRI plays an important role in patients with NF1, as it can delineate dystrophic elements and may also disclose the presence of unrecognized neurofibromas, lateral thoracic meningoceles (see Fig. 23.18), and protrusion of ribs into the thecal sac in patients with dystrophic curves. Curves that appear non-dystrophic on radiographs may be shown to have dystrophic features on MRI. It is important to recognize this, as rapid curve progression is then more likely [45].
Ribs
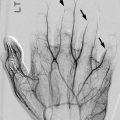
Rib dysplasia occurs in about 60 % of patients with scoliosis and is most marked at the apex of the scoliosis on the convex side. Rib penciling is defined as the rib width being narrower than the narrowest part of the second rib [41]. Ribs may initially appear normal but develop undulations and become irregularly sclerotic and twisted as patients mature. The medial rib ends may appear pointed and if associated with pediculate thinning can protrude into the spinal canal [46]. Dysplasia must be differentiated from notching due to intercostal neurofibromas (Fig. 23.20), which resembles the small, discrete impressions produced by collateral vessels in aortic coarctation.
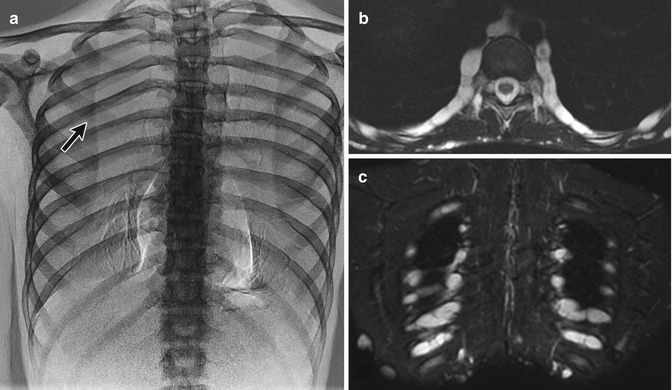
Fig. 23.20
Intercostal neurofibromas in a 16-year-old boy with NF1 (same patient as Fig. 23.15). (a) Dual-energy chest radiograph with bone algorithm shows rib notching at multiple levels, most evident at the posterior fifth right rib (arrow). Axial (a) and coronal (b) T2-W MRI shows multiple hyperintense neurofibromas along the lower edges of the ribs (Courtesy of Sandra L Wootton-Gorges)
Skull and Facial Bones
Macrocephaly is common, with almost half of patients having a skull volume over the 95th percentile. Partial (see Fig. 23.14) or complete absence of the greater or lesser sphenoid wings may lead to pulsating exophthalmos. There may be round or ovoid osseous defects in the lambdoid suture near its bifurcation at the mastoid. Eighty percent of these defects occur on the left; this may be associated with poor aeration of the ipsilateral mastoid air cells. Facial bone dysplasia takes the form of osseous deformity and sclerosis, often with grotesque results. Plexiform neurofibromas may distort facial bones and contribute to development of sphenoid dysplasia (see Fig. 23.14).
Acoustic neuromas, optic gliomas, astrocytomas, ependymomas, meningiomas, and trigeminal nerve tumors are of course assessed by MRI, but mass effect or secondary foraminal enlargement may be evident on radiographs. MRI may also show clinically silent focal areas of increased T2 signal (Fig. 23.15), considered to be areas of heterotopia or hamartoma in the cerebellum, basal ganglia, and other cerebral structures.
Extremities
Fractures are more common in patients with NF1, occurring in bone that appears normal or only slightly osteoporotic, or through an underlying fibrous lesion. Features that suggest neurofibromatosis include segmental osseous sclerosis adjacent to the fracture site, narrowed medullary canal, cystic or hourglass expansion of the medullary canal, and sclerosis and tapering of the ends of the bones at the fracture site. However, fractures with subsequent nonunion can occur in radiographically normal bone.
Nonunion and pseudarthrosis complicate fracture healing in as many as 13 % of patients. Pseudarthrosis may result from dystrophic healing of fractured previously normal bone or follow a pathologic fracture. Histological evaluation of nonunion reveals a hamartomatous mass of connective tissue (not a neurofibroma), and the histology resembles that of non-NF pseudarthrosis or fibrous dysplasia.
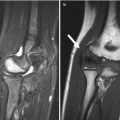
Anterolateral bowing of the tibias is common in NF1 (Fig. 23.21) and may suggest the diagnosis in young children. Fracture is common before age 3 years, and this may be followed by pseudarthrosis of the lower tibia (see Fig. 23.21). Although pseudarthrosis of the tibia can develop in the general population, it is especially common in NF1, occurring in up to 11 % of patients with this diagnosis (conversely, approximately 50 % of patients with tibial pseudarthrosis have NF1). (See Chap. 13 for further discussion of tibial pseudarthrosis.) A similar pathologic process may occur in the fibula, radius, ulna, humerus, clavicle, and other sites. Bowing of the radius with radial head dislocation can be associated with ulnar pseudarthrosis.
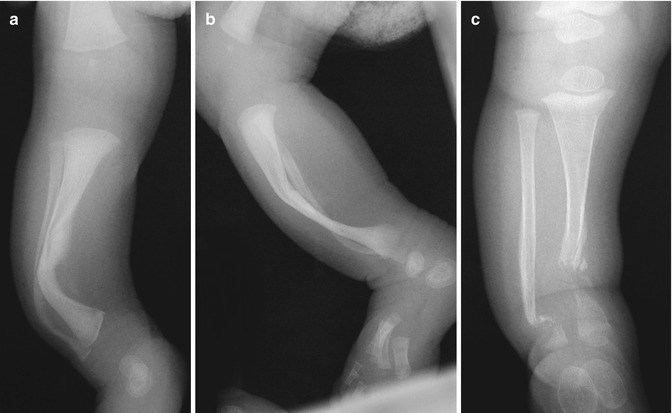
Fig. 23.21
Anterolateral bowing and subsequent pseudarthrosis in a boy with NF1. (a, b) During infancy, there is anterolateral tibial and fibular bowing. (c) Tibial and fibular pseudarthroses develop by about age 2 years (Courtesy of Sandra L. Wootton-Gorges)
Other focal intrinsic and extrinsic osseous abnormalities also occur. Intrinsic fibrous cortical defects and nonossifying fibromas are more common in NF1 than in the general population, and indeed multiple bilaterally symmetric nonossifying fibromas about the knee suggest the presence of NF1 [41]. Multiple nonossifying fibromas accompanied with café-au-lait spots characterize Jaffe-Campanacci syndrome; this may be a manifestation of NF1 (see Chap. 21). Mesodermal dysplasia and intraosseous neurofibromas may be responsible for some cystic lesions (Fig. 23.22). Extrinsic cortical erosions, in contrast, result from pressure caused by adjacent expanding plexiform neurofibromas or localized periosteal thickening. The periosteum in patients with NF1 is abnormally loose, and this can allow extensive subperiosteal hemorrhage.
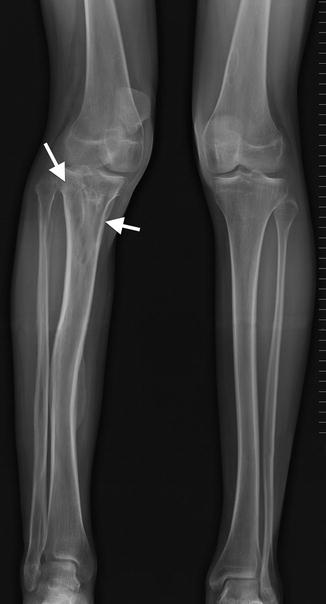
Fig. 23.22
Lucent lesions resembling nonossifying fibromas in the proximal tibia in a patient with NF1. Arrows identify lesions. Dysmorphic growth has resulted in genu valgum (Courtesy of Rebecca Stein-Wexler)
Dysplasia of individual bones may take the form of irregular cortical contour, angular deformity, and irregular cortical sclerosis and lucency. If extreme, sclerosis may resemble melorheostosis. The iliac wing of the pelvis may be enlarged, with a narrow sclerotic base and enlarged obturator foramen. The femoral necks often demonstrate valgus alignment.
Neurofibromatosis can also cause hypertrophy and deformity in limbs. The soft tissues, muscles, and bones are all involved, with an increase in both length and width; plexiform neurofibromas are often present. The resulting massive limb enlargement has been termed “elephantiasis neuromatosa.” This must be differentiated from hemangioma, vascular/lymphatic malformation, and Proteus syndrome.
3 Arthrogryposis
Arthrogryposis constitutes a clinical description (“hooked joint”) rather than a single discrete pathologic entity and is defined as two or more joint contractures in multiple body areas [47]. Approximately 1 in 4,300 live births is affected [47]. Individuals often have reduced or absent skinfolds at involved joints, skin dimples at large joints, adducted shoulder joints, and reduced muscle mass. The condition results from decreased fetal movement, which may be secondary to underlying neurologic or muscle disease, connective tissue or skeletal abnormality, or intrauterine constraint. The clinical features range from nonspecific combinations of joint contractures to well-known, complex syndromes. More than 300 specific disorders exhibit arthrogrypotic features, and a definite diagnosis other than arthrogryposis can be made in approximately 50 % of patients with congenital contractures. As our knowledge of disease genotypes improves, fewer and fewer patients are diagnosed with arthrogryposis.
Arthrogrypotic conditions are generally classified as arthrogryposis multiplex congenita or as classic arthrogryposis (amyoplasia congenital) [48, 49]. Arthrogryposis multiplex congenita is defined as having multiple congenital contractures of all four limbs but can be limited to the upper or lower extremities and can also involve the jaw and trunk. Amyoplasia is more specific: the limbs are affected symmetrically, with the elbows extended, the wrist flexed, camptodactyly, severe equinovarus, knee flexion or extension, and dislocated hips, with no truncal involvement. Cases of arthrogryposis can also be divided into three categories that facilitate diagnosis: group I, only limbs involved; group II, limb and other malformations; group III, limb malformations along with lethality or central nervous system involvement [50]. Patients in groups II and III can often be diagnosed with a specific disorder [48].
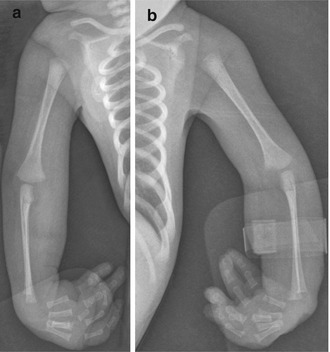
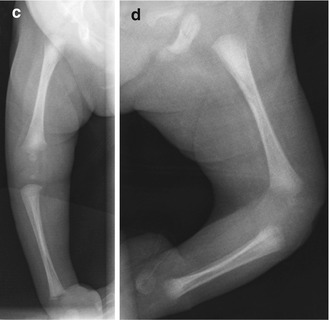
In patients with arthrogryposis, fetal development is initially normal, but joint movement becomes constrained, leading to collagen proliferation, fibrotic replacement of muscle, and marked thickening of joint capsules (the collagenic response). The most extreme form of arthrogryposis is fetal akinesia deformation sequence (Pena-Shokeir syndrome), characterized by—in addition to joint contractures—facial and jaw abnormalities, lung hypoplasia, short umbilical cord, growth retardation, and polyhydramnios.
Arthrogryposis is often found incidentally during routine second trimester ultrasound (US), before the usual maternal perception of fetal movement. However, it may also be identified by third trimester US if the mother reports decrease in fetal movement. Immobilization lasting longer than 3 weeks likely produces pathologic joint contracture, the severity of which is determined by the age at onset and duration of immobility [51]. In the classic picture of arthrogryposis, the newborn is delivered with internally rotated shoulders, extended elbows, pronated forearms, and flexed wrists in ulnar deviation (Fig. 23.23). The scapulae may be non-descended. The hips are usually also flexed, whereas the knees are either flexed or extended. Joint abnormalities are usually bilateral and fairly symmetric, although the degree of contracture is not necessarily equal on both sides (e.g., valgus deformity on one side and varus on the other is rare). The contractures associated with arthrogryposis are severe at birth, and although the underlying abnormalities do not change, untreated deformities worsen as the child grows [52].