Cerebral palsy
Location
Common musculoskeletal imaging abnormalities
Comment
Spine
Long C-shaped thoracolumbar scoliosis with pelvic obliquity, elongated vertebral bodies, increased thoracic kyphosis, lumbar lordosis
Early detection and treatment may prevent severe deformities
Upper extremity
Radial head dislocation, contractures, negative ulnar variance
Uncommon or often overlooked
Hip
Windblown deformity, increased femoral anteversion, coxa valga, subluxation, deformed femoral head
Early acetabular index measurements may predict future dislocation
Knee
Flexion contracture deformity (crouch knee), patella alta, patellar fragmentation, chondromalacia of patella
Foot
Equinovalgus malalignment, hallux valgus
Associated ankle valgus
Cerebral palsy (CP) affects the brain and neurons above the anterior horn cells of the spinal cord; it results in spasticity and difficulty in controlling muscles. The most common cause of physical disability in childhood, it has an incidence of 2.0–2.5 per 1,000 live births [1]. A heterogeneous group of neurodevelopmental disorders, CP causes persistently impaired movement and posture. In 2004, the International Working Group on the Definition and Classification of Cerebral Palsy [2] defined CP as “a group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to nonprogressive disturbances that occurred in the developing fetal or infant brain. The motor disorders of CP are often accompanied by disturbances of sensation, perception, cognition, communication, and behavior; by epilepsy; and by secondary musculoskeletal problems.”
Four major components determine the classification of CP: (1) type and severity of motor abnormalities, (2) anatomical and neuroimaging distribution, (3) associated non-motor impairments, and (4) timing of the presumed causal event (prenatal, perinatal, or postnatal) [3]. A widely used classification system differentiates motor abnormalities according to neuromuscular tone (hypotonia or hypertonia) by spastic (most common, 70 %), ataxic (5–20 %), dyskinetic (dystonia or choreoathetosis, 10–20 %), and mixed forms. The trunk and each of the limbs are assessed separately to define distribution (monoplegia, hemiplegia, diplegia, triplegia, and total body involvement). Although this system is clinically significant, it lacks inter-user reliability [4]. Palisano and colleagues [5] developed the gross motor function classification system (GMFCS), a five-level classification based on the child’s motor abilities, functional limitations, and need for wheel mobility or assistive devices. This has become a simple, intuitive, complementary, and reliable tool to classify gross motor function in children with CP and has been implemented worldwide.
The underlying causes of CP are poorly understood. Several risk factors have been identified, but less is known about their interactions and how they relate to different pathophysiological pathways. A wide range of prenatal, perinatal, and postnatal factors can occur alone or in combination. Common risk factors include prematurity, low birth weight, birth asphyxia, fetal intrauterine exposure to maternal infection and inflammation, maternal fever during labor, multiple gestations, coagulation disorders, pre- or perinatal stroke, maternal thyroid disease, and placental pathology [6]. Recent evidence that CP may be caused by multiple genetic factors will likely influence future therapy [7]. Hypoxic ischemic encephalopathy is the major perinatal cause of neurological morbidity in both premature and full-term newborns. Resulting from asphyxia in the preterm brain, mild to moderate injury results in damage to white matter, manifesting as periventricular leukomalacia or parenchymal hemorrhage; gray matter damage occurs in premature infants with profound hypoxic ischemic encephalopathy or in full-term infants [8].
Spasticity, which typically develops between 6 and 18 months of age, alters previously normal skeletal anatomy [9]. The paraspinal muscles, hip flexors, hip adductors, hamstrings, gastrocnemius, and soleus muscles are most commonly affected. Scoliosis, hip dislocation, and fixed contractures develop during periods of rapid growth and represent the most common progressive manifestations of CP.
In addition, children with CP are at increased risk of fractures due to disuse osteoporosis. Bisphosphonates may be used to increase bone density, reducing fracture risk from 30.6 to 13 % [10]. However, after bisphosphonate therapy fractures may occur at locations where there are large differences in bone density [11]; the transition in density may produce a so-called stress concentration or stress riser, which predisposes to fracture.
Spine
The prevalence of scoliosis in patients with spastic CP ranges from 15 to 61 % [12, 13]. Scoliosis in CP is more common in males and may be painful, whereas idiopathic scoliosis has an eight to one female predominance and is rarely painful. The risk of developing scoliosis increases with GMFCS level and age. Children in GMFCS level IV or V have a 50 % risk of developing moderate or severe scoliosis by 18 years of age, whereas children in GMFCS level I or II have almost no risk [12].
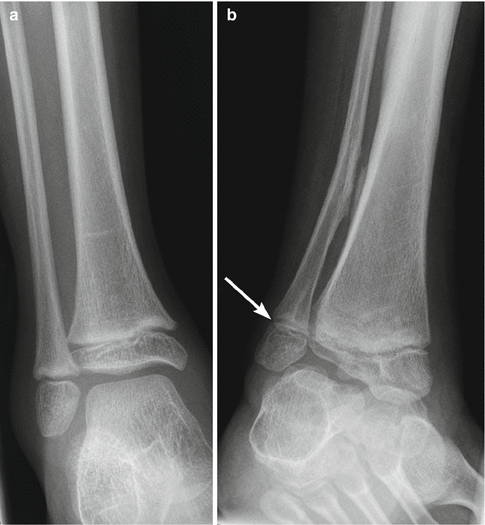
Patients with CP, especially those with spastic diplegia, often suffer from low back pain, which restricts their ability to walk and impairs their quality of life [14]. This may be due to increased thoracic kyphosis (Fig. 24.1) along with lumbar lordosis that results in compression and shearing forces, leading to spondylolysis and spondylolisthesis. Spondylolysis of the fifth lumbar vertebra is found in 21 % of children with CP, four times more often than in normal subjects [14].
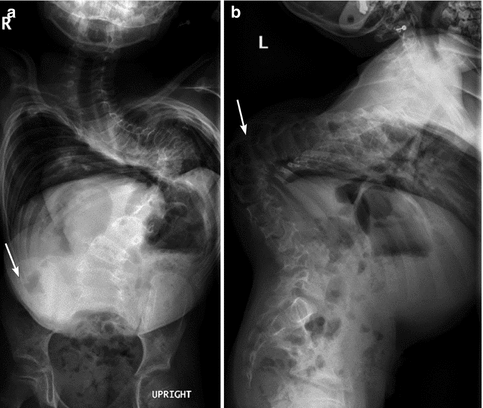
Fig. 24.1
Kyphoscoliosis in an 8-year-old with spastic diplegia. (a) Severe C-shaped levoconvex thoracolumbar scoliosis. The right ribs override the iliac crest (arrow). (b) Marked kyphosis centered at the lower thoracic spine (arrow)
In severely disabled patients, developmental abnormalities of the spine become evident in early childhood, presumably the result of decreased activity, lack of erect weight bearing, abnormal posture, and muscle imbalance. The vertebral bodies are elongated, with increased vertical height and decreased anteroposterior dimensions, resembling those of a dog (Fig. 24.2)—hence the term caninization. The end plates are often convex and the disk spaces small (Fig. 24.3).
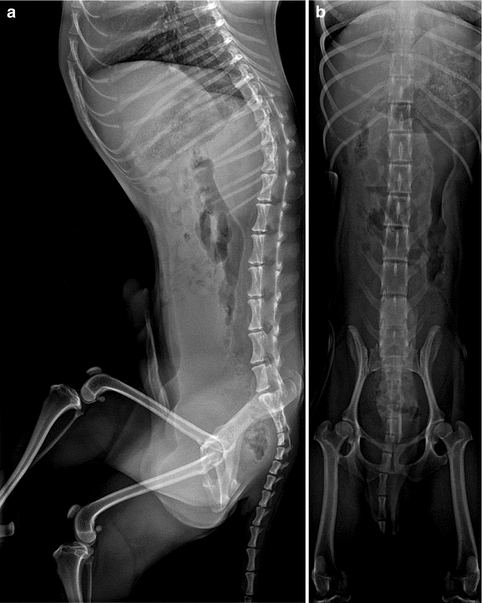
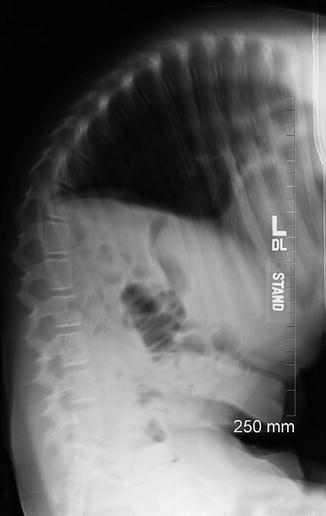
Fig. 24.2
(a, b) Canine vertebrae in a 2-year-old Maltese Terrier. The vertebral bodies are elongated and narrow, similar to those in patients with severe CP who have never born weight (Courtesy of Lynda Zucca)
Fig. 24.3
Caninization of vertebrae in CP. The vertebral bodies are relatively tall, and the intervertebral spaces are narrow and aligned in severe kyphosis, likely due to muscle weakness (Courtesy of Rebecca Stein-Wexler)
In both mild and severe cases, a long C-shaped thoracolumbar curve is most common, followed by a lumbar curve (Fig. 24.4). Most curves evolve from postural to fixed deformities. Progression is greatest during rapid growth in middle adolescence and may continue after skeletal maturity (Fig. 24.5), especially when associated with pelvic obliquity [15]. In patients whose curve is less than 50° at skeletal maturity, researchers have reported an annual progression of 0.8°, whereas the annual progression is 1.4° in those whose curve exceeds 50° at maturity [16].
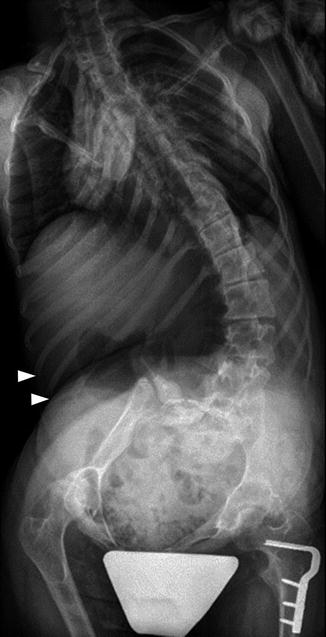
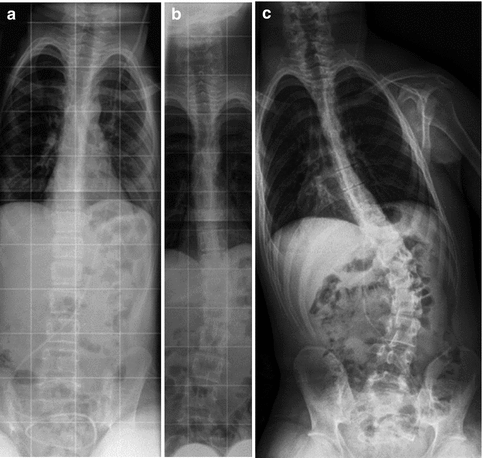
Fig. 24.4
Long C-shaped scoliosis in a 15-year-old girl with CP. The ribs are almost overriding the right iliac bone (arrowheads). Note the “windblown” configuration of the pelvis. Femoral hardware is from the right varus osteotomy
Fig. 24.5
Progressive thoracolumbar scoliosis in CP. Sequential radiographs demonstrate (a) normal alignment at age 13, (b) 19° of scoliosis at age 15, and (c) further progression at age 17
Two thirds of patients with scoliosis have fixed pelvic obliquity, and almost as many have hip subluxation. A characteristic “windblown” configuration with both femurs deviated toward the concave side of the curve may be seen, and in severe cases the ribs may override the iliac crest or even lie within the pelvic brim (see Fig. 24.4).
Treatment
The primary goal of treatment is to improve sitting balance and prevent curve progression. External bracing is poorly tolerated and rarely successful, and therefore, surgical procedures such as anterior release or fusion combined with posterior fusion may be necessary. Correction of increased kyphosis, pelvic obliquity, hip flexion deformity, and hip dislocation is also helpful [17] and contributes to improved quality of life [18].
Upper Extremity
Radial head dislocation is rarely reported, and the incidence of the condition is unknown. Posterior dislocation is most common and is probably caused by combined effects of muscle imbalance, spasticity, and contracture. Elbow, wrist, and finger contractures also occur. Negative ulnar variance can be seen in children with more severe muscle imbalance and tone [19] (Fig. 24.6). If radial head dislocation is painful, excision may be more successful than reconstruction [20].
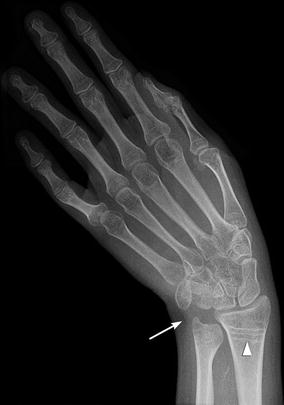
Fig. 24.6
Varus deviation of the hand in a 16-year-old with CP and developmental delay. There is minimal negative ulnar variance (arrow). Note diffuse osteopenia with multiple growth arrest lines in the proximal radial metaphysis (arrowhead)
Lower Extremity
Hip
The prevalence of hip subluxation and dislocation in CP ranges between 3 and 75 % [21]. Progressive hip flexion and adduction lead to windblown deformity (Fig. 24.7), increased femoral anteversion, coxa valga (real or apparent), subluxation, and deformity of the femoral head (Fig. 24.8). Complete hip dislocation is uncommon in patients with CP. Typically, the spastic adductor and iliopsoas muscles induce a twisting or internal rotation of the femur along its longitudinal axis. Thirty to fifty degrees in normal infants, this anteversion should diminish during childhood and measure 8–15° by skeletal maturity.
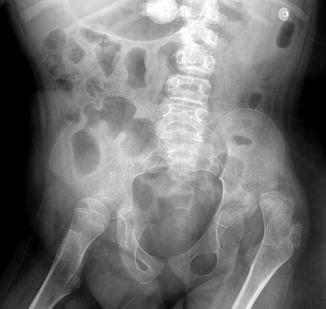
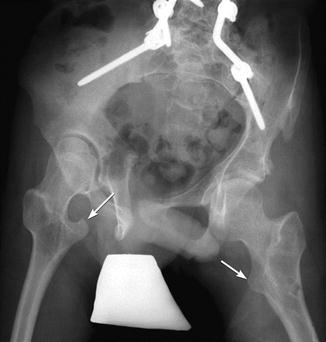
Fig. 24.7
Windblown pelvis in a 10-year-old boy. The right hip is abducted, and the left adducted and also dislocated. Shallow left acetabulum (Courtesy of Tal Laor, Cincinnati Children’s Hospital)
Fig. 24.8
Pelvic deformity in a 16-year-old boy with CP. There is bilateral external rotation and abduction. The pelvis is oblique, with apparent right acetabular protrusion. Prominent lesser trochanters (arrows)
Patients with spastic CP have normal anteversion at birth, but abnormal muscle forces prevent the decrease in torsion that typically occurs after age 2 or 3, causing increased femoral neck anteversion to persist. The anteversion angle averages 55° in children with CP and is even larger in nonambulatory patients [15, 22].
The extent of anteversion is usually assessed clinically, but it can also be calculated from limited magnetic resonance imaging (MRI) [23] or computer tomography (CT) [24, 25], with cuts through the hips and knees. The angle is determined by measuring the angle that the femoral neck makes to a transverse plane through the femoral condyles when the femur is viewed on end [15]. (See Chap. 13 for detailed presentation of measurement technique.)
The femoral neck-shaft angle, measured on a true frontal radiograph, is the angle between the mid-axis of the femoral shaft and a line along the mid-axis of the femoral head and neck. This can be measured on a true frontal radiograph. However, both increased femoral anteversion and improper positioning of the femur can result in projectional foreshortening of the femoral neck, generating the false impression of an increased neck-shaft angle [15]. This should be described as “apparent coxa valga” (Fig. 24.9). In cases of severe CP, valgus may truly be present.
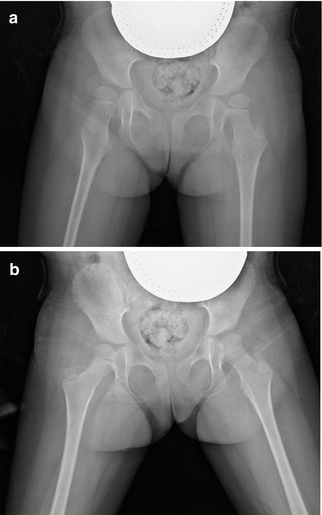
Fig. 24.9
Apparent coxa valga in a 4-year-old girl with CP (GMFCS V). (a) Supine frontal view with hips in external rotation shows apparent coxa valga on the right, with a neck-shaft angle of 180°. (b) Frontal view on the same date with hips internally rotated (and abducted). The femoral neck-shaft angle now appears normal at 160° (Courtesy of Jon R. Davids)
A radiograph with hips internally rotated accurately depicts the neck-shaft angle. Optimal alignment of the proximal femur for radiographic analysis can be assessed by the appearance of the greater and lesser trochanters. The lesser trochanter should not be visualized, whereas the greater trochanteric apophysis should be seen in its entirety. If the lesser trochanter is visualized and the apophysis of the greater trochanter is obscured, then the femur is externally rotated and not properly positioned for accurate assessment of the neck-shaft angle; this positioning results in apparent coxa valga. External rotation of the upper femur causes the lesser trochanter to appear prominent (Fig. 24.10).
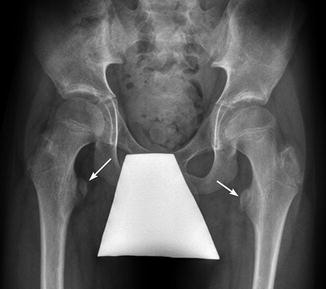
Fig. 24.10
External rotation with increased femoral anteversion in a patient with CP causing valgus appearance of the femoral necks as well as lesser trochanter prominence (arrows)
Femoral head position is initially normal or only slightly subluxated, but severe subluxation is common by age 7 (Fig. 24.11) [26]. Incomplete lateral coverage of the femoral head is not necessarily due to subluxation, but can result from pelvic obliquity, adductor contracture, increased femoral neck anteversion/valgus, or disparity in size between the femoral head and the acetabulum. Furthermore, acetabular dysplasia and secondary degenerative joint disease result from chronic subluxation, and mobility may be impaired [27]. Medial flattening and a triangular or V shape of the femoral head may develop as subluxation progresses (Fig. 24.12) [27]. Abnormal forces may result in irregular ossification of the femoral heads (Fig. 24.13).
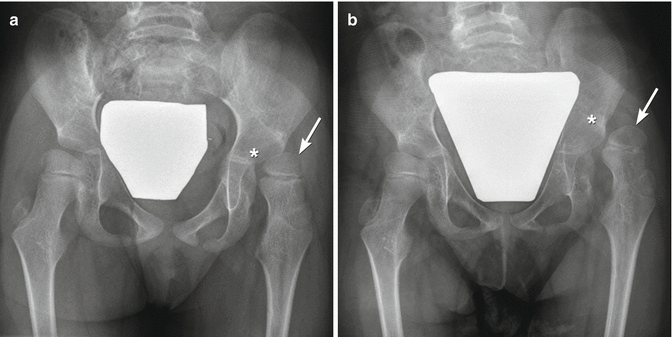
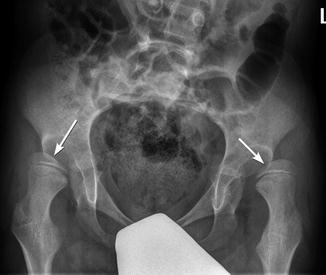
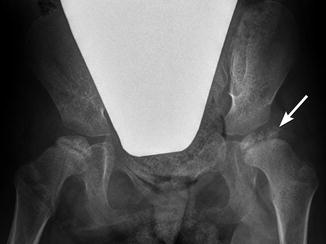
Fig. 24.11
Progressive superolateral migration of the left femoral head in a patient with severe dyskinetic CP. (a) At 3 years old, there is mild superolateral migration of the left femoral head (arrow) and acetabular dysplasia. (b) Three years later, the hip is dislocated (arrow), and a pseudoacetabulum has formed. Bilateral apparent coxa valga
Fig. 24.12
Bilateral superolateral dislocation of the hips in a 9-year-old boy with CP. Note symmetric flattening of the medial aspects of both femoral heads (arrows)
Fig. 24.13
Disturbed ossification of the femoral heads in a 5-year-old patient with spastic quadriplegic CP. Abnormal forces result in irregular ossification of the femoral heads, more pronounced on the left (arrow). However, the overlying cartilage is probably intact
The aim of treatment is to prevent adduction and flexion deformity and progression to subluxation or dislocation [26]. Nonsurgical management entails stretching the spastic agonist muscles, strengthening the weaker antagonist muscles, and splinting in abduction. Surgical procedures include tenotomy. Correction of actual coxa valga and femoral anteversion can be achieved by varus derotation osteotomy.
Knee
Flexion contracture deformity (crouch knee) is the most common knee abnormality in spastic CP. Crouch knee is associated with hip and ankle flexion contractures. Increased force on the quadriceps muscles as well as overstretch of the infrapatellar tendon lead to patella alta, patellar fragmentation, chondromalacia, joint instability, muscle weakness, and pain [28]. Projectional overriding of the femoral condyles and tibial plateau due to fixed flexion contracture may make it difficult to obtain a satisfactory frontal radiograph (See Chap. 11 for discussion of techniques for measuring patella alta.)
Other patellar abnormalities include an elongated or crescent-shaped patella (Fig. 24.14), fragmentation of the lower pole of the patella, chondromalacia, and Osgood-Schlatter-like changes in the tibial tubercle. Hyperextension of the knee (genu recurvatum) is common in spastic CP due to progressive rectus femoris contracture together with abnormally long hamstrings and equinus foot deformity [29].
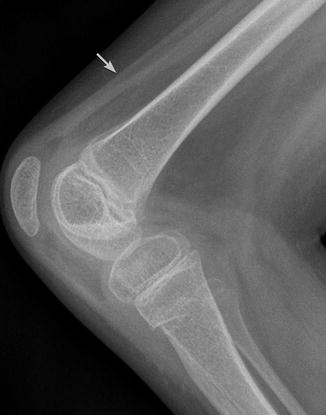
Fig. 24.14
Knee in a 9-year-old boy with paraplegic CP. The patella is elongated and crescent shaped. Bones are osteopenic, and muscles atrophic due to disuse (arrow)
Ankle and Foot
The most common complaint related to foot deformity in children with CP is pain with ambulation or use of orthoses. Abnormal shoe wear patterns are often evident. Foot deformities usually result from imbalance between the extrinsic muscles of the lower leg that control foot and ankle alignment [30]. This may be secondary to spasticity, disrupted motor control, and/or impaired balance. Typically, the ankle plantar flexors (gastrocnemius and soleus) are overactive, and the ankle dorsiflexors are ineffective.
In children with CP, there are three common coupled foot and ankle segmental malalignment patterns: equinus, equinoplanovalgus, and equinocavovarus. In addition, there are two common secondary malalignments, ankle valgus and hallux valgus, which may be associated with any of the three segmental malalignment patterns. (See Chap. 15 for more detailed discussion of these abnormalities, as well as specific information on measurement techniques.) Standardized radiographic analysis of foot deformity in these children should include three views: frontal and lateral views of the foot, along with a frontal view of the ankle. Although it is difficult to obtain weight-bearing projections in severely disabled children, they are necessary for standardization and later comparison.
Equinus at the Ankle
Equinus results from fixed spastic contracture of the gastrocnemius and soleus, causing the characteristic tiptoe or toe-heel gait. Equinus is commonly associated with knee flexion and valgus or varus deformity of the hindfoot and forefoot.
Equinovalgus Malalignment
This is characterized by equinus deformity of the hindfoot, coupled with pronation deformity of the midfoot and forefoot. As the contracture worsens, the calcaneus becomes displaced posterolaterally and everts, allowing the talus to drop into a vertical position (Fig. 24.15), with eventual rocker-bottom deformity (increased talocalcaneal angle) (Fig. 24.16). Ankle valgus and hallux valgus deformities are frequently associated with equinovalgus foot. Equinovarus is less common but can be present in up to 20 % in patients with spastic CP.
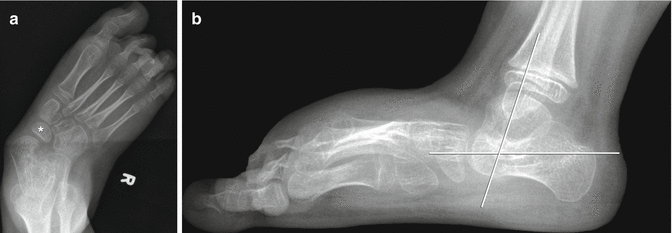
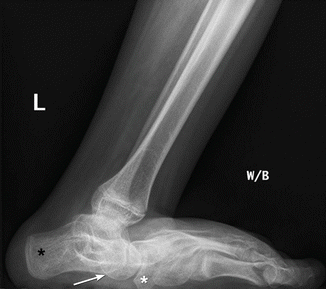
Fig. 24.15
Foot deformity in a patient with spastic CP. (a) There is hindfoot and forefoot valgus with the talus directed medial to the base of the first metatarsal. The navicular is laterally displaced relative to the talus. (b) The talus is almost vertical, with its long axis almost parallel to that of the tibia. The talocalcaneal angle is increased
Fig. 24.16
Rocker-bottom deformity in a 13-year-old boy with spastic CP. Lateral weight-bearing view shows posterior displacement of the calcaneus (black asterisk) and vertical talus (arrow) resulting in rocker-bottom deformity. The navicular is displaced downward by the vertical talus. The tibia and fibula are inclined anteriorly with respect to the long axis of the foot. Note fatty infiltration of the muscles
Ankle Valgus
This is determined by the tibiotalar angle, assessed on a frontal weight-bearing ankle radiograph (Fig. 24.17). Ankle valgus is often corrected when it contributes to hindfoot valgus deformity. The goal of treatment of ankle and foot deformities is to increase function by preventing and correcting deformities.
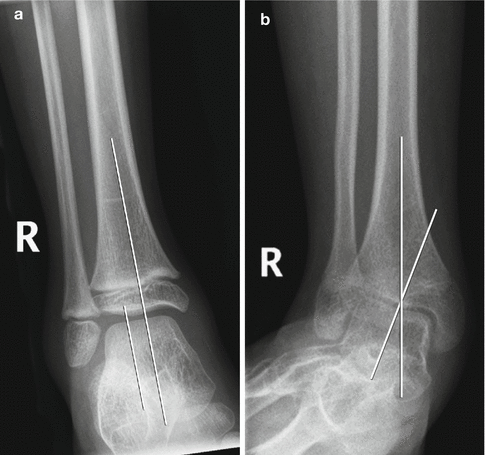
Fig. 24.17
Ankle valgus deformity in a 13-year-old boy with spastic diplegia. (a) Normal ankle for comparison, with the long axis of the talus parallel to that of the tibia. (b) There is lateral tilt of the talus with respect to the long axis of the tibia
Treatment
Early management of spasticity with pharmacologic injections (e.g., Botulinum toxin), neurosurgery (selective dorsal rhizotomy, intrathecal baclofen), and application of orthotics helps prevent the development of fixed deformities of the muscle-tendon unit. Musculoskeletal surgeries include tendon transfer to address dynamic muscle imbalance and osteotomies/arthrodeses to fix malalignment that cannot be corrected simply by manipulation [15, 30].
2 Spinal Cord/Motor Unit Lesions
Spinal cord/motor unit lesions constitute a group of disorders that affect nerve fibers traveling from the spinal cord to the muscles. They include developmental disorders of the spinal cord (neural tube defects), spinal cord trauma, and hereditary and infectious disorders of the anterior horn cells (spinal muscular atrophy, poliomyelitis).
Flaccid paralysis, which is accompanied by muscle atrophy, helps identify lower motor neuron defects. Neural tube defects, such as myelomeningocele, are among the most common disorders requiring imaging prior to orthopedic management. Although myelomeningocele is often characterized as a typical lower motor neuron lesion, it is frequently accompanied with upper motor neuron signs, including spinal cord reflex activity and spasticity. Loss of proprioception and sensation challenges bony stabilization procedures in patients with myelomeningocele.
2.1 Developmental: Neural Tube Defects (Dysraphism)
Spinal dysraphism encompasses a group of congenital anomalies caused by defective neural tube closure and anomalous differentiation of the caudal cell mass. Neural tube defects refer to a wide range of abnormalities that result from the maldevelopment of the neuropore and the adjacent mesodermal and ectodermal structures during embryogenesis (see Chap. 3 for further discussion of spine development). The defects are classified as open or closed according to the presence or absence of exposed neural tissue. Open spina bifida (“spina bifida aperta”) is caused by the failure of primary neurulation, resulting in exposed neural tissue or meninges with or without cerebrospinal fluid leakage. It includes two main types: myelomeningocele and meningocele. Myelomeningocele is a severe form in which the neural tissue and the meninges protrude through an opening in the spine, whereas with meningocele only the meninges protrude. Incidence in males and females is equal.
Spina bifida occulta is caused by defects in secondary neurulation and includes diastematomyelia, diplomyelia, dorsal dermal sinus, spinal lipoma, and isolated bone defects. In these forms, skin covers the neural elements, but there may be overlying cutaneous lesions such as nevi or depigmentation, hairy patches, hemangiomas, dimples, and subcutaneous lipomas. Occult lesions are more common in boys than in girls [31].
Neural tube defects result from multifactorial genetic, environmental, and combined factors. Chromosome malformations are present in a small minority of individuals, most frequently trisomy 13 or 18. Maternal diabetes, folic acid deficiency, and anticonvulsant drugs are a few common environmental risk factors that may cause neural tube defects; with widespread use of antenatal screening and folic acid dietary supplementation, the incidence has fallen [32].
Prenatal diagnosis of open spina bifida is common in countries with advanced medical care and is based on ultrasound (US) evidence of associated cerebral signs, including ventriculomegaly, scalloping of the frontal bones, and abnormal curvature of the cerebellar hemispheres. Evaluation of the spine may show absence of the dorsal arches or interrupted skin contour with or without meningocele and splaying of the laminae (Fig. 24.18). The antenatal diagnosis of closed spina bifida is difficult since cranial signs are absent and diagnosis relies only on direct evidence of the lesion, hydrosyringomyelia, or a low-lying cord—all difficult to identify by prenatal US although they are readily apparent at MRI (Fig. 24.19).
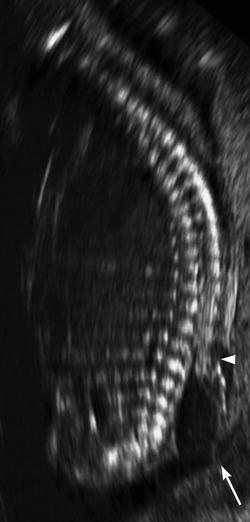
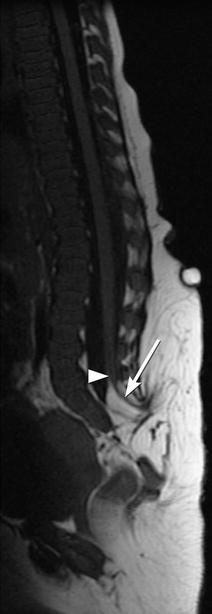
Fig. 24.18
Prenatal sagittal US at 17 weeks gestational age shows a meningocele sac (arrow) and a defect in the posterior elements of the spine (arrowhead) (Courtesy of Felipe Moretti)
Fig. 24.19
Newborn with lipomyeloschisis, a type of skin-covered spina bifida. Sagittal T1-W MRI shows a low-lying cord (arrowhead) reaching the sacral level and connecting with deep extension of a subcutaneous lipoma through spinal dysraphism (arrow)
Myelomeningocele (Box 24.2) accounts for more than 98 % of open spinal dysraphisms [33]. Up to 85 % of myelomeningocele patients have coexisting hydrocephalus, and nearly all have an associated Chiari II malformation [34] (Fig. 24.20).
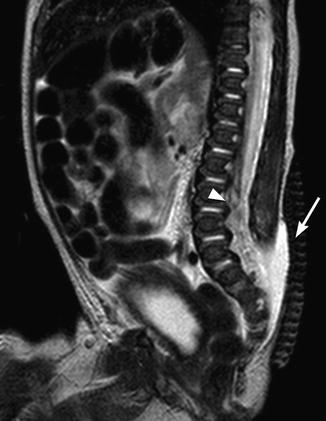
Fig. 24.20
Newborn with a lumbosacral myelomeningocele. A sagittal T2-W MRI shows complete absence of the midline posterior cartilage, muscle, and skin layers at the level of the open spinal dysraphism (arrow). The distal spinal cord is low lying (arrowhead). There was also a Chiari II malformation of the brain (not shown)
Box 24.2: Common Musculoskeletal Imaging Abnormalities in Myelomeningocele
Myelomeningocele | |
|---|---|
Location | Common musculoskeletal imaging abnormalities |
Spine | Scoliosis: congenital or acquired |
Kyphosis: congenital or acquired | |
Developmental lordosis | |
Hip | Subluxation and dislocation |
Coxa valga | |
Contractures | |
Knee | Extension and flexion contractures deformities |
Foot | Equinus deformity |
Calcaneus foot | |
Valgus deformity | |
Clubfoot | |
Vertical talus | |
The child with myelomeningocele needs intensive support from a multidisciplinary team that includes radiologists, neurosurgeons, orthopedic surgeons, urologists, neurologists, and rehabilitation therapists. Bone abnormalities associated with myelomeningocele may be congenital or acquired, specific to myelomeningocele or similar to deformities seen in other conditions. Most of these deformities occur in childhood. Acquired abnormalities are caused by paralysis, muscle imbalance, bone malformation, or a combination of factors. The level of dysraphism may cause weakness or paralysis of agonist muscle groups and spasticity of antagonist groups, leading to dynamic muscle imbalance, disrupted musculoskeletal growth, and functional impairment.
All children with myelomeningocele have some degree of tethered cord, and this typically persists or recurs after surgical release due to scarring or adhesions. This tethering of the neural elements may not cause problems, but pain, decreased function in the lower extremities, and progressive scoliosis may be clinical signs of a significant tethering lesion. Tethered cord should be suspected as a cause of scoliosis in children younger than 6 years old with a high lumbar lesion.
Imaging
Prenatal imaging often begins with US. Fetal MRI is complementary, providing superior anatomic resolution regardless of maternal body habitus or fetus position. Fetal MRI facilitates recognition of the craniocervical abnormalities of Chiari II malformation and delineation of less common types of dysraphism (Fig. 24.21), such as terminal myelocystocele [35] or lipomeningocele [36]. It also provides the opportunity to evaluate congenital scoliosis (Fig. 24.22).
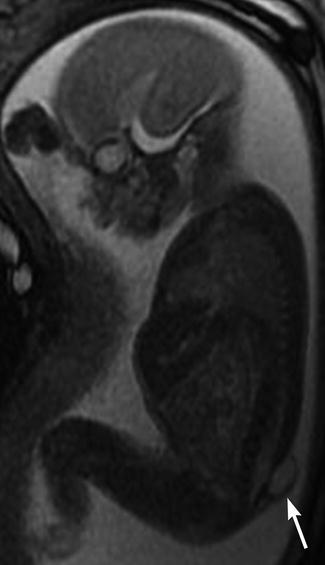
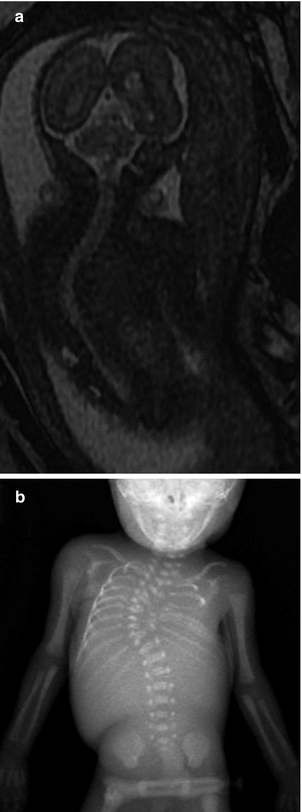
Fig. 24.21
Meningocele in a 23-week-old fetus. Sagittal FIESTA (fast imaging employing steady-state acquisition) MRI shows a lumbosacral defect and meningocele (arrow)
Fig. 24.22
Thoracic scoliosis in a 22-week-old fetus. (a) Coronal FIESTA MRI shows dextroscoliosis. (b) Postmortem radiograph shows fixed scoliosis with multiple vertebral segmentation anomalies and chest deformity (Courtesy of Julie Hurteau-Miller)
Postnatal imaging should include cranial and renal ultrasound, which can be performed after surgical correction of dysraphism. Severe kyphosis may displace the psoas muscles anteriorly, which pushes the kidneys together so they may abut each other at the midline, putting them at risk during surgery. In addition, cross-sectional imaging may show that the aorta is unusually short, and it may have an abnormal branching pattern. Brain MRI is especially important in the neonate with brainstem-related symptoms, as it allows assessment of the brainstem and craniocervical junction. Presurgical spine radiographs are helpful, and spine MRI may be needed if the clinical features are atypical or to help with surgical decision-making.
Radiographs are helpful later in life to assess scoliosis, hip position, and other associated bone abnormalities. If there is spinal deformity, spine radiographs should be performed annually, but rapidly progressive curves or curves associated with neurological manifestations should be imaged with MRI of the brain and spine [37]. Supine frontal and lateral spine radiographs allow assessment of structural dysraphism and hip position, but as the child matures, upright views are essential to demonstrate the effect of gravity.
The frontal projection demonstrates fusiform increase in interpediculate distance, with the widest separation at the midsegment; this point also corresponds to the apex of the kyphosis (if present). Disk spaces are narrowed and vertebral bodies appear to be widened, although measurement may not confirm this. The spinous processes are absent and laminae cannot be identified, as they are forced outward by the expanding meningocele and therefore project anteriorly and laterally (Fig. 24.23). The transverse processes project forward and the pedicles laterally. The facets may be fused into a continuous mass that is initially cartilaginous and later osseous (Fig. 24.24). In infants, the normal anterior vertebral body notch is often absent at the defect level.
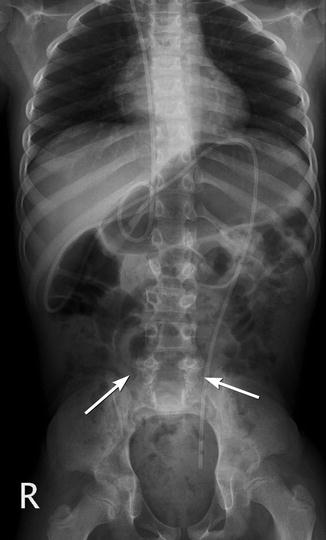
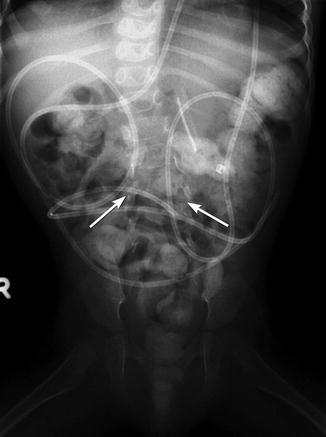
Fig. 24.23
Spinal dysraphism, L5 and sacrum (arrows). Note absent spinous processes from L5 to S5. Ventriculoperitoneal shunt
Fig. 24.24
Lumbosacral dysraphism. The vertebrae are wide, and the interpediculate distance is increased. There are no identifiable lumbar laminae or spinous processes, and the pedicles project laterally (arrows). Note fecal retention, gastrostomy tube, and ventriculoperitoneal shunt tubing, all associated findings in patients with meningomyelocele
Scoliosis
Spinal curvature is common, including scoliosis, kyphosis, and lordosis. These deformities can be congenital or acquired. Congenital curves are less common, present in only about 15 % of children, and associated with hemivertebrae or bony bars. They are unlikely to progress. Most curves are acquired, caused by mechanical instability; these tend to increase during childhood, as paresis progresses [38, 39]. The progressive nature of these deformities may cause severe disability and interfere with both rehabilitation and ambulation. A review of the predictors of scoliosis in patients with myelomeningocele found that most curves develop early in life (Fig. 24.25) [40], about 40 % of curves begin to form after age 9, and a few develop later, up to about 15 years of age.
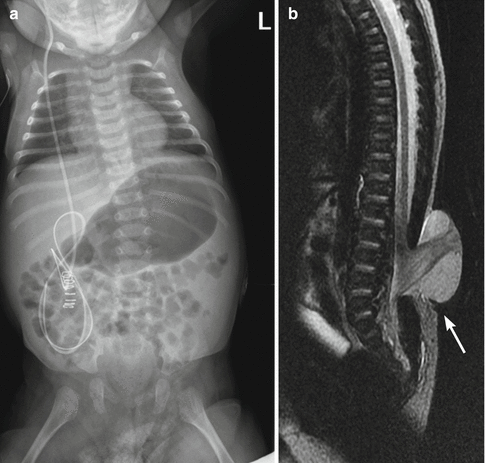
Fig. 24.25
Newborn with myelomeningocele and congenital scoliosis. (a) There is mild thoracolumbar C-shaped levoscoliosis. (b) Sagittal T2-W MRI shows low-lying cord, lumbar dysraphism, and herniation of the meninges and neural tissue (arrow), fusing with neural placode
Clinically significant scoliosis in patients with myelomeningocele is defined as a curve with a Cobb angle measuring more than 20°, because smaller curves often fluctuate, resolve, or even reverse direction [40]. Children with a thoracic neurological injury level have a 90 % incidence of scoliosis [41], with curves often greater than 45°. The lower the dysraphic defect, the less the incidence of scoliosis. Only 10 % of those with dysraphism at or below L4 will develop scoliosis.
Scoliosis in children with myelomeningocele can be attributed to muscle weakness due to high paraplegia, asymmetric paralysis, or spastic hemiplegia due to hydrocephalus. This usually causes a C-shaped scoliosis with associated kyphosis (Fig. 24.26). S-shaped scoliosis can occur in children with hydromyelia or hydrosyringomyelia with hydrocephalus [42]; shunting may halt the progression of neurological deficits, but usually does not result in recovery of function, and scoliosis may still progress. Scoliosis may be accompanied by pelvic obliquity, which may compromise seating balance and lead to ischial pressure sores.
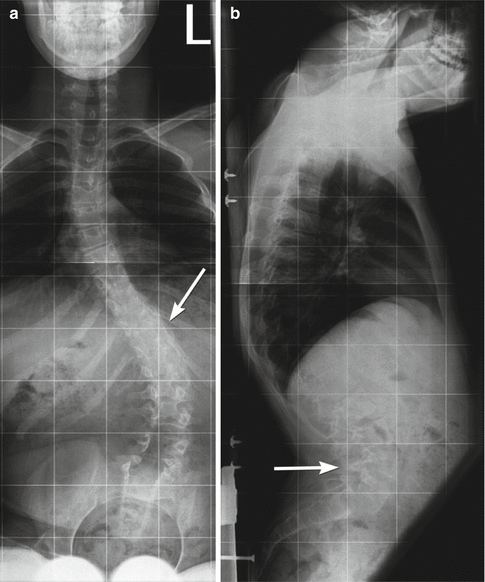
Fig. 24.26
Kyphoscoliosis in a 14-year-old boy with myelomeningocele. (a) The neural arch defect begins at about T10 (arrow). (b) Thoracic kyphosis is centered at this level, and there is also exaggerated lumbar lordosis (arrow)
Increasing scoliosis should prompt assessment for hydrocephalus, shunt-related complications, syrinx, and recurrent tethering of the cord (Fig. 24.27). Recurrent tethering at the site of the initial neural canal closure is quite common [43]. MRI of the brain and spine depicts most of these entities, but re-tethering of the cord can be difficult to assess and requires clinical correlation. CT or limited MRI can be used to follow hydrocephalus.
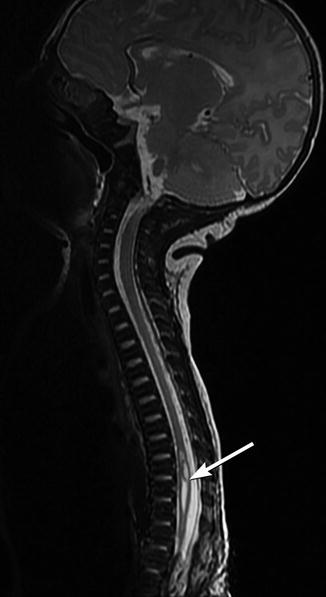
Fig. 24.27
Newly developed syrinx in a child with repaired myelomeningocele and increasing scoliosis. Sagittal T2-W MRI shows a cavity (arrow) in the spinal cord
Large curves may cause pain, make sitting in a chair difficult, and restrict pulmonary function (Fig. 24.28). If scoliosis continues to progress (Fig. 24.29) after neurological problems have been corrected, orthopedic treatment should be considered, as symptoms may improve following spinal realignment with fusion [44]. Conservative orthotic treatment is relatively ineffective for curve correction. Spinal fusion with correction of scoliotic deformity can improve pulmonary function and sitting balance as well as prevent further progression of the curve. Careful planning for spinal surgery is required as children with myelomeningocele are at increased risk of wound infections, pseudarthrosis, and progression of deformity.
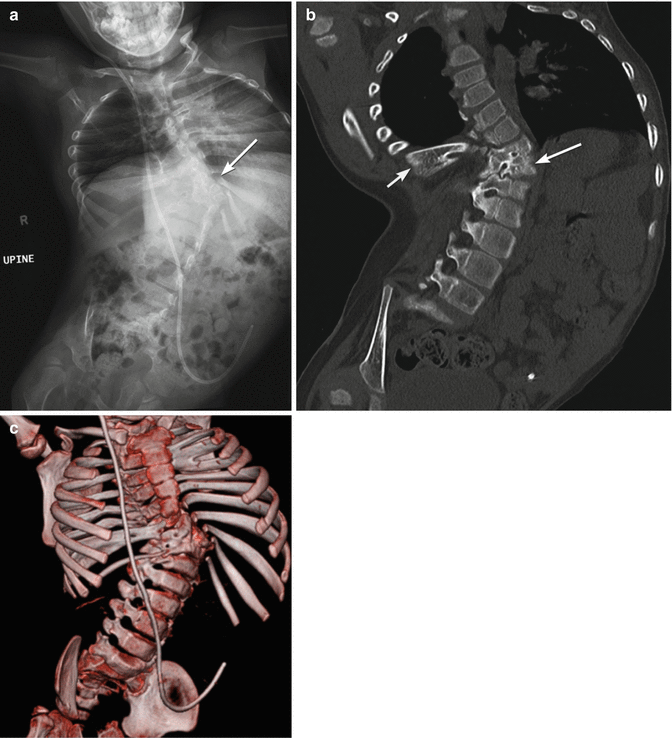
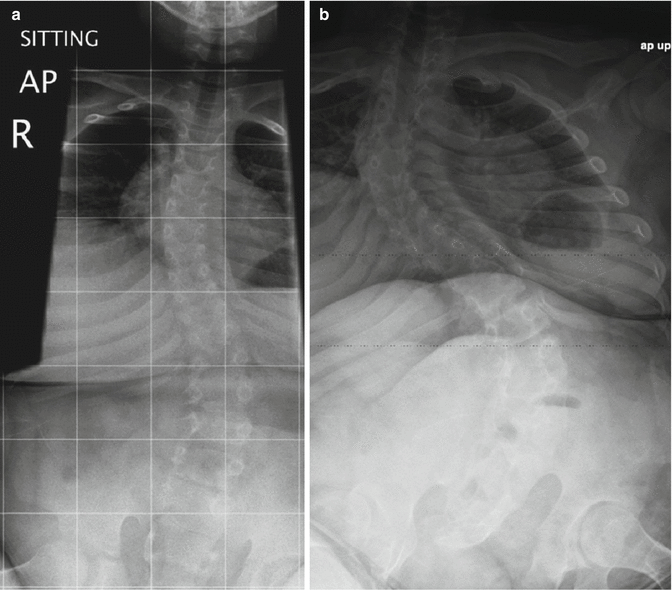
Fig. 24.28
Spina bifida and multiple segmentation abnormalities resulting in small lung volumes in a 5-year-old boy. Radiograph (a), coronal CT reconstruction (b), and 3-D CT reformatted image (c) demonstrate dysraphism extending from the mid-thoracic spine distally, with fused vertebrae on the right as well as multiple hemivertebrae on the left at the apex of the scoliosis (arrow, (a); long arrow, (b)). Ribs are fused (short arrow, (b)) Additional fused ribs at the upper left chest. Note dislocated right hip
Fig. 24.29
Progressive S-shaped scoliosis in a boy ((a), 9 years old; (b), 15 years old) with spina bifida, sacral agenesis, tethered cord, and cloacal exstrophy
Kyphosis
Kyphosis usually occurs in the lower thoracic and upper lumbar spine, with an incidence of 10–46 %. Lumbar kyphosis at birth can make closing the skin and meningeal defects difficult. Later, progression of the kyphosis can lead to breathing difficulties because crowded abdominal structures press upward on the diaphragm. Children also have difficulty feeding, since they often lean forward and must therefore use their hands to support themselves; this may lead to failure to thrive [45]. Treatment is not easy but should be initiated early to increase abdominal space, avoid pressure on the diaphragm and lungs, and improve truncal balance.
Congenital kyphosis is characterized by a severe angular deformity with a prominent gibbus at the apex of the curve. Paralytic kyphosis is more common than the congenital form and is classified as collapsing kyphosis or rigid-sharp angle kyphosis. Collapsing kyphosis is often C shaped and initially flexible. The apex may occur anywhere in the lower thoracic or lumbar spine (see Fig. 24.26). Rigid sharp-angle kyphosis is more common in older children and is usually S shaped with an accompanying upper thoracic lordosis. All kyphotic curves tend to progress [42, 46, 47]. Vertebrae are wedged or bullet shaped in lateral projection.
Developmental lordosis with or without scoliosis is usually compensatory, counteracting instability caused by pelvic obliquity or a dislocated hip. In the past, this deformity was associated with lumboperitoneal shunting, but this method of shunting is rarely used today [48].
Hip and Lower Extremity
Hip Subluxation
Deformity and instability of the hip in myelomeningocele is common, ranging from joint contracture to subluxation and dislocation. The presence of hip deformity and dislocation is related to the neurological level of the disorder and resultant muscle imbalance. Nerves for the hip flexors and adductors originate at L1–L3, whereas innervation of the hip abductors and extensors originates at L5–S2. The myelo-hip can be divided into three groups, based upon the level of the neurological injury. The first is thoracic/high lumbar level, in which none of the muscles about the hip are significantly innervated, and dislocation develops slowly over time due to flaccid paralysis. The second is low lumbar level, in which the hip flexors, hip adductors, and knee extensors are functionally innervated, whereas the hip extensors, hip abductors, and knee extensors are flail. Hip subluxation and dislocation occur rapidly due to muscle imbalance. The final group is injured at the sacral level. In this group, all muscles about the hip are functionally innervated, and subluxation/dislocation is unusual.
Subluxation and dislocation are very common in children with myelomeningocele, occurring in up to 50 %. In most cases, hips are normal at birth and become dislocated or dysplastic after the first year of life. However, infants who were in the breech position or those who are placed on their side after surgical closure of dysraphism frequently develop early dislocation. Hip US allows early diagnosis in these cases, but because most patients with myelomeningocele develop dysplasia or dislocation more than 1 year after birth, radiographs are in general most useful [38] (Fig. 24.30). The same measurements apply in myelomeningocele as in developmental dysplasia of the hip (Chap. 11).
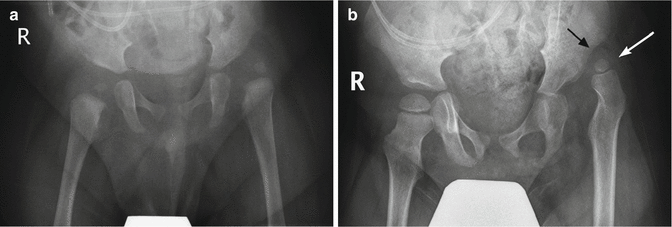
Fig. 24.30
Progressive hip dislocation in patient with myelomeningocele. (a) At 1 year of age, the left hip is dislocated and the acetabulum dysplastic. Note sacral defect. (b) Two years later, the degree of dislocation has increased (white arrow), and a pseudoacetabulum is present (black arrow). The femoral head is smaller than on the normal side. Bilateral coxa valga
Pavlik harness and closed reduction are used to treat hip dysplasia or instability in preambulatory patients with a sacral level of neurological impairment. However, due to the underlying muscle imbalance, reduction is usually followed by recurrent dislocation. Treatment of hip deformity in older children depends on the neurological level, the specific deformity, and the potential for ambulation. Reduction of a dislocated hip may involve both femoral and acetabular surgery.
Hip Flexion
Hip flexion deformity may be seen in patients with thoracic or lumbar defects and is frequently associated with abduction contracture, which results from unusually tight tensor fascia lata. When unilateral, there is also pelvic obliquity and scoliosis. Surgery is directed at release of hip muscles.
Femoral Torsion
In children with myelomeningocele who have abnormal gait and activity levels, femoral torsion fails to decrease normally as the child grows. This results in inward rotation at the knees and in-toeing of the legs and feet, as in patients with CP [38] (see Chap. 13 for discussion of measurement techniques). Both flexion and extension contractures may develop at the knee, interfering with ambulation, upright position, and orthotic fitting. Flexion contractures are probably the most common knee deformity, and contractures greater than 30° often impair the ability to walk. Extension contractures are infrequent and most often encountered in children who were in breech presentation, as intrauterine compression can lead to hyperflexed hips, hyperextended knees, and clubfoot. Extension contractures may also result from extensive immobilization.
Rotational Deformities
Rotational deformities of the lower extremities are common in both ambulatory and nonambulatory children with myelomeningocele. An in-toeing gait is usually dynamic if secondary to medial hamstring dominance or fixed if secondary to internal tibial torsion. A short fibula and a weak soleus muscle can lead to excessive lateral twisting of the tibia, resulting in external tibial torsion and an out-toeing gait. Selective fibular shortening is common in children with myelomeningocele. Normally, by age 4 the distal fibular growth plate should be at the level of the talar dome, and it should be even lower in older children. In many patients with myelomeningocele, the distal fibular physis is at or above the level of the distal tibial physis (Fig. 24.31). A short distal fibula causes ankle valgus deformity. This combined with external tibial torsion results in abnormal loading of the ankle, leading to a wedge-shaped distal tibial epiphysis, which is narrower laterally. This deformity contributes to planovalgus foot malalignment, which can compromise standing and use of orthoses.