8 Skeletal Anomalies Self-Help Organization Title: Little People of America (English/Spanish) Description: Provides mutual support to people of short stature (4 feet 10 inches/1.5 m and under) and their families. Information on physical and developmental concerns, employment, education, disability rights, medical issues, adaptive devices, etc. Newsletter. Provides educational scholarships and medical assistance grants, access to medical advisory board, assistance in adoption. Local, regional, and national conferences and athletic events. Online chat room. Scope: National Number of groups: 54 chapters Founded: 1957 Address: P.O. Box 745, Lubbock, TX 79408, United States Telephone: 1–888-LPA-2001 E-mail: LPADataBase@juno.com Definition: Fatal skeletal dysplasia with a marked shortening of the torso and extremities and a comparatively large head. Incidence: 0.2–0.5/10 000 births. Sex ratio: M : F = 1 : 1. Clinical history/genetics: Partly autosomal-recessive inheritance. Autosomal-dominant inheritance is also known (new mutations). Teratogens: Not known. Origin: In isolated cases, a defect in synthesis of collagen type II has been proved. Ultrasound findings: There are various forms of achondrogenesis (type I and type II). Severe shortening of the extremities, a narrow thorax and reduced ossification of the vertebral column and the skull are characteristic features. In some cases, hygroma colli or fetal hydrops may also be present. Thickening of the soft tissue of the arms is typically seen. Ascites and fetal hydrops may also develop. The abdomen and head are very large compared to the chest and the limbs. In contrast to osteogenesis imperfecta, the skull is not compressible, and fractures of the long bones are not a feature; fractures of the ribs may sometimes be seen. The diagnosis is possible as early as 12 weeks of gestation, due to the appearance of a thickened nuchal fold (nuchal swelling) and skeletal malformations. Clinical management: Molecular-genetic diagnosis, further ultrasound screening including fetal echocardiography. In cases of achondrogenesis, cardiac anomalies are seen rarely in comparison with other skeletal disorders. Radiographic examination of the skeletal system leads to further confirmation. Termination of pregnancy has to be considered, as the outcome is fatal. Management of the pregnancy and labor should not be influenced by fetal distress. Procedure after birth: As the fetus is not viable, intensive medical interventions are not recommended. References Benacerraf B, Osathanondh R, Bieber FR. Achondrogenesis type 1: ultrasound diagnosis in utero. JCU J Clin Ultrasound 1984; 12: 357–9. Gabrielli S, Falco P, Pilu G, Perolo A, Milano V, Bovicelli L. Can transvaginal fetal biometry be considered a useful tool for early detection of skeletal dysplasias in high-risk patients? Ultrasound Obstet Gynecol 1999;13:107–11. Glenn LW, Teng SS. In utero sonographic diagnosis of achondrogenesis. JCU J Clin Ultrasound 1985; 13: 195–8. Johnson VP, Yiu CV, Wierda DR, Holzwarth DR. Midtrimester prenatal diagnosis of achondrogenesis. J Ultrasound Med 1984; 3: 223–6. Mahony BS, Filly RA, Cooperberg PL. Antenatal sonographic diagnosis of achondrogenesis. J Ultrasound Med 1984; 3: 333–5. Ozeren S, Yüksel A, Tom T. Prenatal sonographic diagnosis of type I achondrogenesis with a large cystic hygroma [letter]. Ultrasound Obstet Gynecol 1999; 13: 75–6. Sauer I, Klein B, Leeners B, Cotarelo C, Heyl W, Funk A. [Lethal osteochondrodysplasias: prenatal and postnatal differential diagnosis; in German.] Ultraschall Med 2000; 21: 112–21. Soothill PW, Vuthiwong C, Rees H. Achondrogenesis type 2 diagnosed by transvaginal ultrasound at 12 weeks’ gestation. Prenat Diagn 1993; 13: 523–8. Tongsong T, Srisomboon J, Sudasna J. Prenatals diagnosis of Langer–Saldino achondrogenesis. J Clin Ultrasound 1995; 23: 56–8. Wenstrom KD, Williamson RA, Hoover WW, Grant SS. Achondrogenesis type II (Langer–Saldino) in association with jugular lymphatic obstruction sequence. Prenat Diagn 1989; 9: 527–32. Won HS, Yoo HK, Lee PR, et al. A case of achondrogenesis type II type. associated with huge cystic hygroma: prenatal diagnosis by ultrasonography. Ultrasound Obstet Gynecol 1999; 14: 288–90. Definition: This is the most frequent heterozygous, nonfatal type of skeletal dysplasia (the homozygous form is fatal), with severe shortening of the limbs and a large head (dwarfism). Adult height: 116– 140 cm. Incidence: 0.5–1.5 per 10 000 births. Sex ratio: M: F = 1 : 1. Clinical history/genetics: Autosomal-dominant inheritance. New mutations are seen in 80%. Affected gene: FGFR3, gene locus: 4 p 16.3. Teratogens: Not known. Pathogenesis: Reduced chondral ossification. Mutation of the fibroblast growth factor receptor. In the homozygous form, early manifestation and fatal outcome. In the heterozygous form, a normal ultrasound appearance is possible up to 20 weeks. Ultrasound findings: Disproportionate hyposomia (dwarfism) with short limbs, large head, and a typical facial profile—protruding forehead (also known as frontal bossing) and flattened nasal bridge. The shortening of the bones of the extremities is apparent in the second half of pregnancy. The measurement of the long bones lies well below the fifth percentile, with accentuation of the proximal part. The hands and feet appear short and stubby. Hydramnios develops in the last trimester. The diagnosis is made with certainty after 24 weeks of gestation, the most reliable quotient being femur length to biparietal diameter, as the femur is often only minimally shortened, but the head is typically very large (macrocrania). In some cases, ventriculomegaly is also observed. Differential diagnosis: Asymmetrical growth restriction, trisomy 21, hypochondroplasia, Kniest syndrome, Russell–Silver syndrome, Shprintzen syndrome, spondyloepiphyseal dysplasia, Turner syndrome. Clinical management: Further ultrasound screening including fetal echocardiography, karyotyping, molecular-genetic diagnosis. There is a danger of cervical column compression due to narrowing of the foramen magnum. Thus, any clinical intervention that may cause manipulation in the neck region during labor and delivery, such as forceps or vacuum extraction, poses a high risk of complications. For this reason, a cesarean section may be an option for delivery. Procedure after birth: Radiography of the skeletal system confirms the diagnosis. Hydrocephalus may develop secondary to occlusion of the foramen magnum. Surgical techniques involving lengthening of the bones of the extremities can lead to an increase in body height of about 20 to 25 cm. Prognosis: Life expectancy in these children is normal. Intelligence is not restricted. Neurological complications, especially involving the cervical spine, are common. In homozygous cases (both parents affected), the outcome is fatal—stillbirth or neonatal death resulting from hypoplasia of the lungs. References Chitayat D, Fernandez B, Gardner A, et al. Compound heterozygosity for the achondroplasia–hypochondroplasia FGFR3 mutations: prenatal diagnosis and postnatal outcome. Am J Med Genet 1999; 84: 401–5. Gabrielli S, Falco P, Pilu G, Perolo A, Milano V, Bovicelli L. Can transvaginal fetal biometry be considered a useful tool for early detection of skeletal dysplasias in high- risk patients? Ultrasound Obstet Gynecol 1999; 13: 107–11. Huggins MJ, Smith JR, Chun K, Ray PN, Shah JK, Whelan DT. Achondroplasia–hypochondroplasia complex in a newborn infant. Am J Med Genet 1999; 84: 396–400. Kurtz AB, Filly RA, Wapner RJ, et al. In utero analysis of heterozygous achondroplasia: variable time of onset as detected by femur length measurements. J Ultrasound Med 1986; 5: 137–40. Lemyre E, Azouz EM, Teebi AS, Glanc P, Chen MF. Achondroplasia, hypochondroplasia and thanatophoric dysplasia: review and update [review]. Can Assoc Radiol J 1999; 50: 185–97. Mesoraca A, Pilu G, Perolo A, et al. Ultrasound and molecular mid-trimester prenatal diagnosis of de novo achondroplasia. Prenat Diagn 1996; 16: 764–8. Modaff P, Horton VK, Pauli RM. Errors in the prenatal diagnosis of children with achondroplasia. Prenat Diagn 1996; 16: 525–30. Moeglin D, Benoit B. Three-dimensional sonographic aspects in the antenatal diagnosis of achondroplasia. Ultrasound Obstet Gynecol 2001; 18: 81–3. Schlotter CM, Pfeiffer RA. Prenatal ultrasonic diagnosis of achondroplasia. Ultraschall Med 1985; 6: 229–32. Definition: Asymmetrical disruptive anomalies, with amputated limbs and splitting defects such as ventral wall defects. The cause is thought to be amniotic bands resulting from premature rupture of the amnion. Incidence: One in 1300 births. Sex ratio: M : F = 1 : 1. Clinical history/genetics: Occurrence mostly sporadic. Rarely associated with congenital defects of fibrous tissue development, as in Ehlers-Danlos syndrome and epidermolysis bullosa. Pathogenesis: The extent of malformations depends on the time at which the disturbances occur. Thus, anencephaly, encephalocele, facial clefts, abdominal wall defects and ectopia cordis may result. Later disturbance causes amputation of extremities and fusion of fingers, as in syndactyly. Ultrasound findings: The findings are extremely variable, affecting many fetal structures. In mild forms, isolated fingers or toes may be missing. Club feet and malposition of the hands may be seen. Localized swellings of the distal parts of the limbs have also been observed (“constriction ring”). On scanning, amniotic bands can be detected in the amniotic cavity. Amniotic bands have to be differentiated from adhesions within the uterine cavity, which may also have connections to the amniotic cavity and are covered with amnion and chorion. These are often a result of curettage, but are not responsible for fetal malformations. In addition to malformations of the limbs, other severe defects are observed in amniotic band syndrome: abdominal wall defects, encephalocele, facial clefts, micrognathia, multiple anomalies of body surface (limb–body stalk anomaly, probably related in its pathogenesis to amniotic band syndrome). Differential diagnosis: Gastroschisis, chromosomal aberrations, club feet, hypoplasia of femur, subcutaneous lymphangioma, Proteus syndrome, Klippel–Trenaunay–Weber syndrome, neural tube defect, omphalocele, Beckwith–Wiedemann syndrome, pentalogy of Cantrell. Clinical management: Further ultrasound screening, including fetal echocardiography and karyotyping. Prognosis: This depends on the severity of the malformations. References Abuhamad AZ, Romero R, Shaffer WK, Hobbins JC. The value of Doppler flow analysis in the prenatal diagnosis of amniotic sheets. J Ultrasound Med 1992; 11: 623–4. Berlum KG. Amniotic band syndrome in second trimester associated with fetal malformations. Prenat Diagn 1984; 4: 311–4. Chen CP. First-trimester sonographic demonstration of a mobile cranial cyst associated with anencephaly and amniotic band sequence. Ultrasound Obstet Gynecol 2001; 17: 360–1. Daly CA, Freeman J, Weston W, Kovar I, Phelan M. Prenatal diagnosis of amniotic band syndrome in a methadone user: review of the literature and a case report. Ultrasound Obstet Gynecol 1996; 8: 123–5. Fiske CE, Filly RA, Golbus MS. Prenatal ultrasound diagnosis of amniotic band syndrome. J Ultrasound Med 1982; 1: 45–7. Herbert WN, Seeds JW, Cefalo RC, Bowes WA. Prenatal detection of intra-amniotic bands: implications and management. Obstet Gynecol 1985; 65: 36S–38S. Hill LM, Kislak S, Jones N. Prenatal ultrasound diagnosis of a forearm constriction band. J Ultrasound Med 1988; 7: 293–5. Jabor MA, Cronin ED. Bilateral cleft lip and palate and limb deformities: a presentation of amniotic band sequence? [review]. J Craniofac Surg 2000; 11: 388–93. McGuirk CK, Westgate MN, Holmes LB. Limb deficiencies in newborn infants. Pediatrics 2001; 108: E64. Merz E, Gerlach R, Hoffmann G, Goldhofer W. Amniotic band syndrome in ultrasound. Geburtshilfe Frauenheilkd 1984; 44: 576–8. Pedersen TK, Thomsen SG. Spontaneous resolution of amniotic bands. Ultrasound Obstet Gynecol 2001; 18: 673–4. Quintero RA, Morales WJ, Phillips J, Kalter CS, Angel JL. In utero lysis of amniotic bands. Ultrasound Obstet Gynecol 1997; 10: 316–20. Wehbeh H, Fleisher J, Karimi A, Mathony A, Minkoff H. The relationship between the ultrasonographic diagnosis of innocent amniotic band development and pregnancy outcomes. Obstet Gynecol 1993; 81: 565–8. Definition: This is a heterogeneous group of disorders all of which have multiple joint contractures present at birth. These may be caused by connective-tissue, muscular, or neurological abnormalities. Incidence: One in 3000–10 000 births. Sex ratio: M : F = 1 : 1. Clinical history/genetics: Autosomal-dominant, autosomal-recessive or X-linked inheritance have all been observed. The distal forms are more likely to occur in families, but sporadic occurrence is also possible. Teratogens: Hyperthermia, perinatal infections, mother suffering from myasthenia gravis. Classification and ultrasound finding: The classification of arthrogryposis is still disputed. A primary condition is known, in addition to secondary, symptomatic disorders (of neuromuscular or central nervous system origin, mainly due to infections). Three basic forms of arthrogryposis are identified: 1, only the extremities are affected; 2, a generalized neuromuscular disorder is present; 3, the central nervous system is involved. The limbs are typically fixed in a certain position; the legs may be stretched or bent, the arms are bent. “Clenched fists” may be observed. The feet are stretched or appear clubbed. Hypoplasia of muscular tissue is evident. Fetal movement is restricted or totally absent. Swelling of the limbs is an accompanying feature. In some cases, the disorder first becomes evident as late as in the third trimester. Hydramnios is seen frequently. In 10% of the cases, anomalies of the central nervous system such as agenesis of the corpus callosum, lissencephaly, ventriculomegaly, or aplasia of the cerebellar vermis are associated. Associated syndromes: Over 120 syndromes are known to occur concomitantly with arthrogryposis, such as Freeman–Sheldon syndrome, multiple pterygium syndrome, Pena–Shokeir syndrome, Smith–Lemli–Opitz syndrome, trisomy 18, Larsen syndrome, trisomy 8 mosaic. Fig. 8.2 Arthrogryposis multiplex congenita. Fig. 8.3 Arthrogryposis multiplex congenita. Fig. 8.5 Arthrogryposis multiplex congenita. Fig. 8.6 Arthrogryposis multiplex congenita. Fig. 8.8 Arthrogryposis multiplex congenita. Fig. 8.9 Arthrogryposis multiplex congenita. Clinical management: Further ultrasound screening, including fetal echocardiography and karyotyping. Search for infections (TORCH). A fixed breech position presents frequently, which may lead to complications during labor and delivery. Procedure after birth: This is determined after an exact diagnosis is made. Prognosis: This depends on the associated malformations and the severity of the disorder and can vary from a fatal outcome to minimal orthopedic handicap. Self-Help Organization Title: Avenues: A National Support Group for Arthrogryposis Description: Connects families affected by arthrogryposis with each other for mutual support and sharing of information. Educates medical and social-service professionals. Semiannual newsletter $10/year. Scope: National network Founded: 1980 Address: P.O. Box 5192, Sonora, CA 95370, United States Telephone: 209–928–3688 (PST) E-mail: avenues@sonnet.com References Ajayi RA, Keen CE, Knott PD. Ultrasound diagnosis of the Pena–Shokeir phenotype at 14 weeks of pregnancy. Prenat Diagn 1995; 15: 762–4. Baty BJ, Cubberley D, Morris C, Carey J. Prenatal diagnosis of distal arthrogryposis. Am J Med Genet 1988; 29: 501–10. Bui TH, Lindholm H, Demir N, Thomassen P. Prenatal diagnosis of distal arthrogryposis type I by ultrasonography. Prenat Diagn 1992; 12: 1047–53. Degani S, Shapiro I, Lewinsky R, Sharf M. Prenatal ultrasound diagnosis of isolated arthrogryposis of feet. Acta Obstet Gynecol Scand 1989; 68: 461–2. Dudkiewicz I, Achiron R, Ganel A. Prenatal diagnosis of distal arthrogryposis type 1. Skeletal Radiol 1999; 28: 233–5. Falsaperla R, Romeo G, Di Giorgio A, Pavone P, Parano E, Connolly AM. Long-term survival in a child with arthrogryposis multiplex congenita and spinal muscular atrophy. J Child Neurol 2001; 16: 934–6. Goldberg JD, Chervenak FA, Lipman RA, Berkowitz RL. Antenatal sonographic diagnosis of arthrogryposis multiplex congenita. Prenat Diagn 1986; 6: 45–9. Gorczyca DP, McGahan JP, Lindfors KK, Ellis WG, Grix A. Arthrogryposis multiplex congenita: prenatal ultrasonographic diagnosis. JCU J Clin Ultrasound 1989; 17: 40–4. Madazli R, Tuysuz B, Aksoy F, Barbaros M, Uludag S, Ocak V. Prenatal diagnosis of arthrogryposis multiplex congenita with increased nuchal translucency but without any underlying fetal neurogenic or myogenic pathology. Fetal Diagn Ther 2002; 17: 29–33. O’Flaherty P Arthrogryposis multiplex congenita [review]. Neonatal Netw 2001; 20: 13–20. Yau PW, Chow W, Li YH, Leong JC. Twenty-year follow-up of hip problems in arthrogryposis multiplex congenita. J Pediatr Orthop 2002; 22: 359–63. Definition: Skeletal dysplasia with short limbs, club foot, swelling of the ears, and progressive joint and spinal column deformities. Incidence: Rare. Sex ratio: M : F = 1 : 1. Clinical history/genetics: Autosomal-recessive inheritance, single-gene heredity. Teratogens: Not known. Ultrasound findings:
General information
Achondrogenesis
Achondroplasia
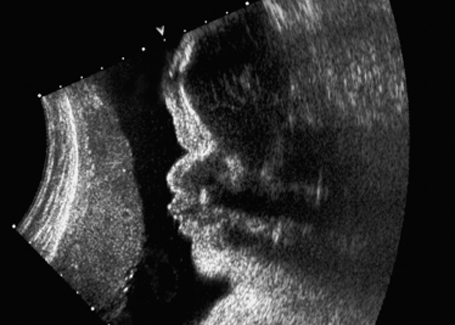
Amniotic Band Syndrome
Arthrogryposis Multiplex Congenita (Multiple Congenital Contractures)
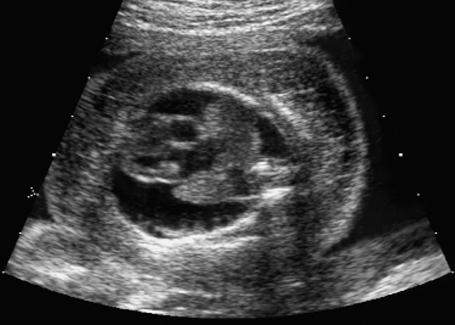
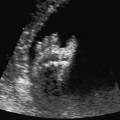
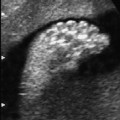
Cross-section of the fetal thorax at 21 + 6 weeks, with arthrogryposis sequence: hydrothorax, severe subcutaneous edema (anasarca).
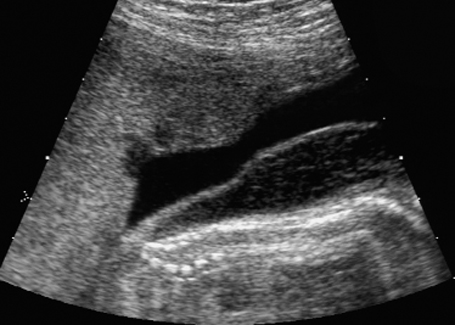
Dorsoanterior longitudinal section of a fetal thorax in fetal arthrogryposis sequence at 21 + 6 weeks, showing severe anasarca.
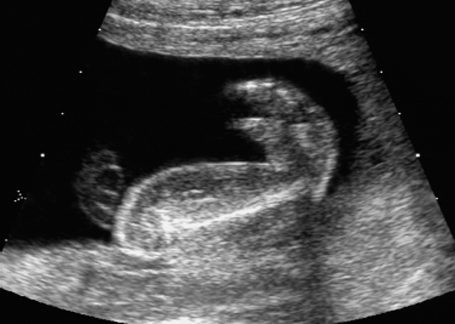
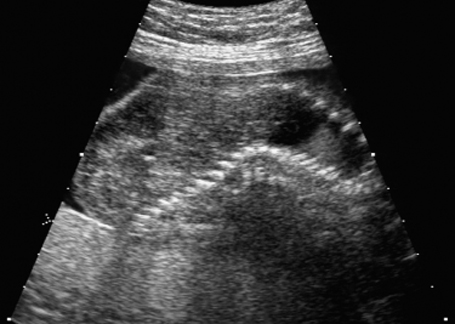
Deformation of the vertebral column in fetal arthrogryposis sequence at 21 + 6 weeks.
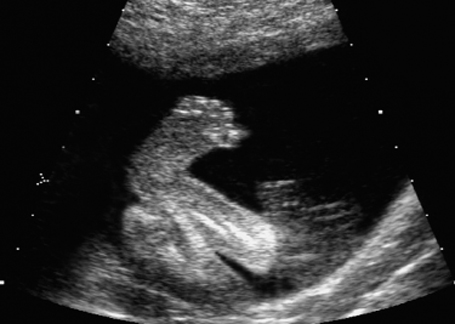
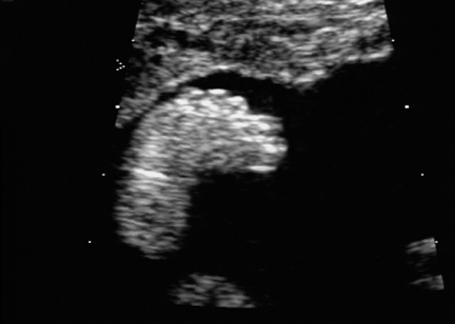
Club foot in fetal arthrogryposis sequence at 21 + 6 weeks.
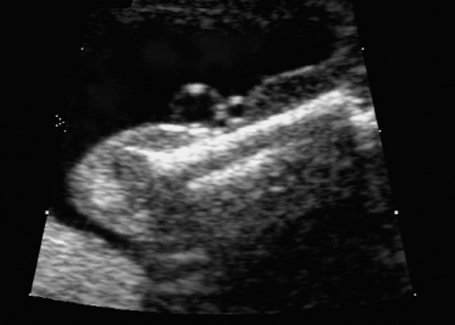
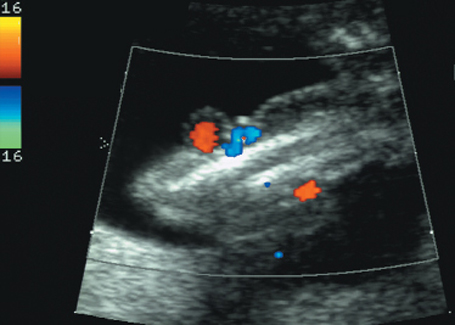
Same case as before. Doppler color mapping shows coils of umbilical cord.
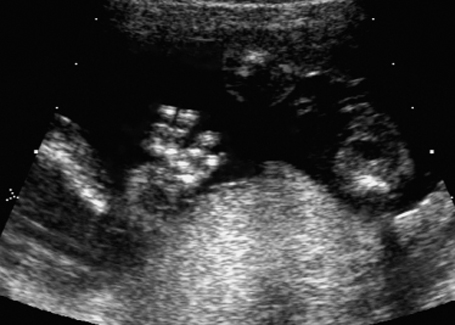
Malpositioning of the fetal hand in arthrogryposis multiplex congenita at 18 + 3 weeks.
Diastrophic Dysplasia
![]()
Stay updated, free articles. Join our Telegram channel


Full access? Get Clinical Tree


