Outline
Embryology, 346
Embryonic Phase, 349
Pseudoglandular Phase, 349
Canalicular Phase, 349
Saccular Phase, 349
Alveolar Phase, 349
Key Features for Assessment, 349
Pulmonary Hypoplasia, 350
Congenital Pulmonary Airway Malformation, 351
Bronchogenic Cyst, 354
Bronchial Atresia, 355
Congenital Lobar Emphysema, 357
Congenital High Airway Obstruction Syndrome, 357
Congenital Hydrothorax, 359
Congenital Diaphragmatic Hernia, 362
Summary of Key Points
- •
The most common chest masses are congenital pulmonary airway malformations (CPAMs) and congenital diaphragmatic hernias (CDHs). For both, the displacement of the heart from its normal position is the most commonly recognized ultrasound finding.
- •
CPAMs are unilateral 98% of the time and appear as echogenic areas of the lung, with or without identified cysts. The CPAM volume ratio (CVR) can be used to subdivide these malformations into high- and low-risk categories for development of fetal hydrops.
- •
Fluid-filled dilated airways should raise suspicion for bronchial or laryngeal atresia. Bilateral hyperechogenic lungs with fluid-filled airways are characteristic of congenital high airway obstruction syndrome (CHAOS), whereas a unilateral hyperechogenic lung with fluid-filled airways suggests bronchial atresia or congenital lobar emphysema (CLE).
- •
Hydrothorax may be primary or secondary. Primary hydrothorax is usually chylous in origin and may progress to hydrops, usually associated with significant skin edema. A large unilateral hydrothorax, even if associated with significant hydrops, may respond to prenatal drainage and shunting.
- •
CDHs are left-sided and posterior in 85% of cases. They are associated with other anomalies (structural and chromosomal) in 40% to 50% of cases.
- •
Prognosis for CDHs depends on the presence of associated abnormalities, the gestational age at delivery, and the degree of pulmonary hypoplasia.
- •
Estimation of lung volume by two-dimensional ultrasound lung-head ratios or volumes obtained through magnetic resonance imaging (MRI) or three-dimensional ultrasound may help guide counseling.
The neonatal identification of chest lesions frequently relies on decompensation of the newborn. Prenatal identification allows for appropriate triaging for site of delivery as well as early and appropriate treatment. Better definition and prenatal identification of chest lesions along with enhanced understanding of the natural history of these lesions has resulted in improvements in counseling. In addition, prenatal therapies are now available for many different types of chest abnormalities. The appropriate use of these therapies depends on accurate identification and assessment of the lesion and the associated pathologic changes. Identifying prognostic factors facilitates continued improvement in the understanding of the natural course of the disease.
This chapter will explore the range of chest lesions that can be identified prenatally and will discuss factors associated with a thorough sonographic diagnosis to facilitate complete and accurate counseling for patients.
Embryology
Although an intact pulmonary system is not required for intrauterine viability, the prenatal development of the respiratory system is integral to ex utero life and survival. To allow successful transition to neonatal life, fetal lungs must proceed through both structural and functional maturation processes, which occur from the early embryonic period and continue after birth. During structural development, there is branching of the airways and development of alveolar spaces, allowing gas exchange to occur once the first breath is taken at delivery. Functional development results in the creation of a surfactant system. This system is composed of phospholipids that decrease alveolar surface tension, inhibiting alveolar collapse during exhalation. The surfactant system develops in the third trimester and is typically mature at 36 weeks’ gestation. Birth prior to the development of the surfactant system or completed pulmonary development results in neonatal respiratory compromise.
The respiratory diverticulum (lung bud) appears as an outgrowth of the ventral wall of the foregut. The epithelium of the internal lining of the larynx, trachea, bronchi, and lungs is entirely of endodermal origin. The cartilaginous, smooth muscle, and connective tissues of the trachea and lungs are derived from splanchnic mesoderm that surrounds the tubular framework. The lung bud is in open communication with the foregut. The diverticulum develops in a craniocaudal direction, leading to development of the upper respiratory structures (nose and pharynx) prior to the lower structures. Two tracheoesophageal ridges separate the diverticulum from the foregut. The dorsal portion of the foregut divides into the esophagus and the ventral portion into the trachea and lung buds.
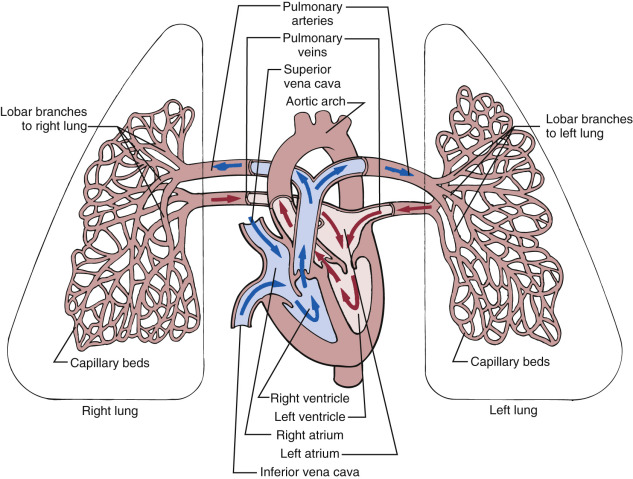
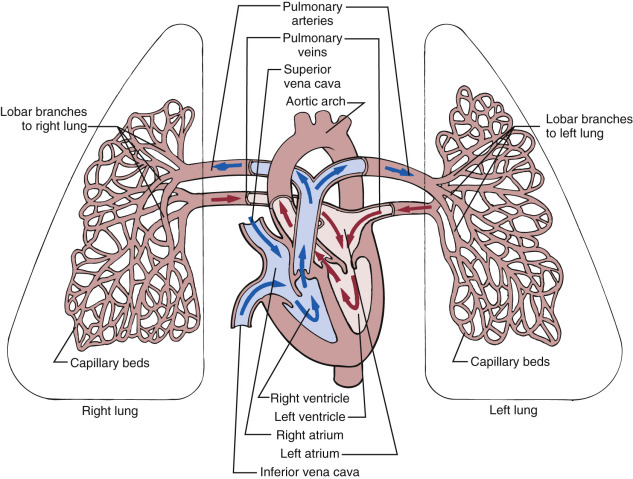
The pulmonary circulation is a highly specialized vascular network, connecting the interdependent heart and lungs ( Fig. 12-1 ). The vascular bed parallels the airways and links the arterial and venous poles of the heart. The pulmonary vasculature has been described as appearing de novo within the mesoderm ventral and lateral to the foregut endoderm, suggesting that this mesoderm pool gives rise to the future pulmonary vasculature as lung develops from the foregut. The lungs have two sets of lymphatic vessels, a superficial set located beneath the pleura and a deep set which follows the blood vessels and extends along the bronchi. Both sets end within the bronchial glands. Efferent lymphatic vessels travel up the trachea, ending at the left-sided thoracic duct or the right-sided lymphatic duct. The fetal lungs play a key role in amniotic fluid volume maintenance: 15 mL/kg of body weight in fluid is produced by the lungs and flows out via the trachea and mouth, circulating and contributing to the amniotic fluid that is swallowed.
The basic structure of the diaphragm is also established early in gestation. Initially, the septum transversum develops, lying caudal to the heart and rostral to the umbilicus, in a position that eventually divides the intraembryonic cavity into the pleuropericardial cavity and the peritoneal cavity. The diaphragm subsequently undergoes programmed muscularization; this represents the final stage in the formation of the basic foundation of the diaphragm and is completed by 14 weeks’ gestation.
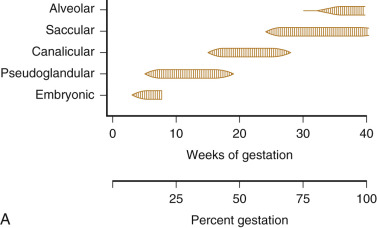
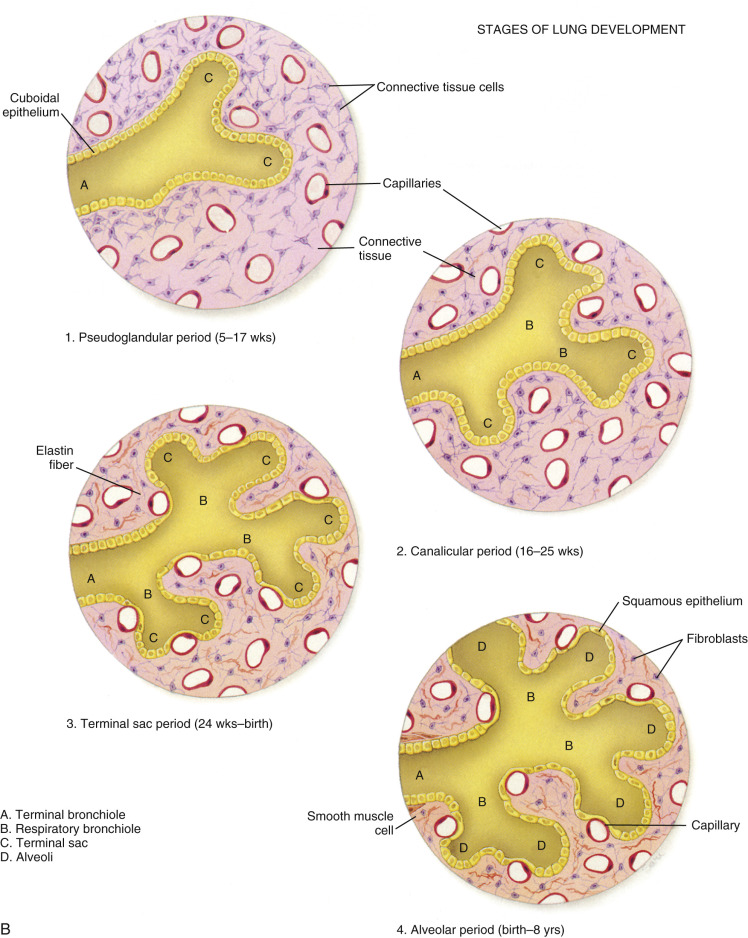
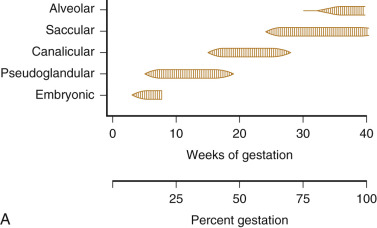
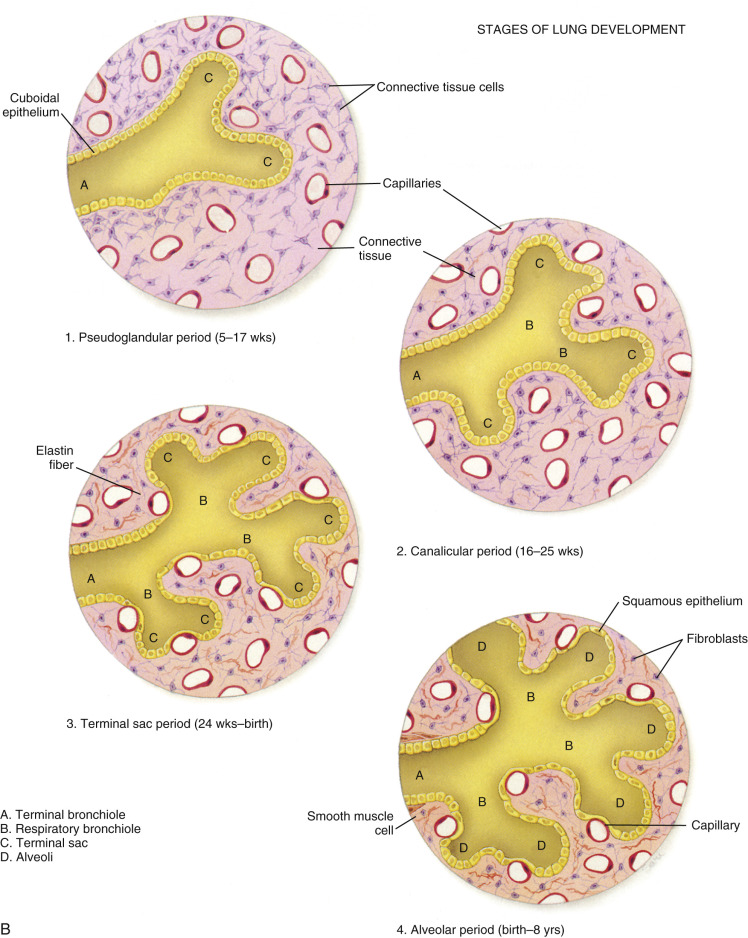
Fetal lung development occurs in five stages: embryonic, pseudoglandular, canalicular, saccular, and alveolar ( Fig. 12-2 ). The timing of these phases is approximate. Transitions between stages are gradual, with overlap from one stage to the next.
Embryonic Phase
The embryonic phase of lung development takes place during the first 4 to 5 weeks of gestation. During this phase, the larynx, trachea, and lung bud form from the foregut. The embryonic phase of lung development begins at about day 28 with the formation of a groove in the ventral lower pharynx, the sulcus laryngotrachealis. A few days later, at about day 30, a bud forms from the lower part, creating the true lung primordium. Development progresses in the 8-week-old embryo as the lobar buds subdivide and form the bronchopulmonary segments. Early on, the asymmetry of the main bronchi is established, with the smaller bud on the left directed more laterally than the larger one on the right, which is parallel to the esophagus and directed more caudally. The embryonic phase proceeds with the unequal dividing of the endodermal branches followed by further division. At the end of the embryonic period, the five lobes of the lungs (three right and two left) are present.
Pseudoglandular Phase
The pseudoglandular stage takes place between the 7th and 16th weeks of embryonic development. Conducting airways are formed by progressive branching of the original lung buds into smaller and more numerous areas. Each bud eventually becomes an independent respiratory unit, served by a bronchiole surrounded by capillary vessels that will bring blood to the lungs for oxygen. During this stage, the first differentiation of lung epithelium occurs. By 13 weeks’ gestation, cilia appear in the proximal airways.
Canalicular Phase
Lasting until approximately 25 weeks’ gestational age, the canalicular phase is crucial for the development of the gas-exchanging portion of the lung. By 20 weeks, there is differentiation into type I pneumocytes, the primary structural cell of the alveolus. Capillaries grow in close proximity to the distal surface of the alveolar cells. Lamellar bodies develop in type II alveolar cells and are the site of surfactant storage prior to release into the alveoli. A sufficient differentiation of the type II pneumocytes into the type I pneumocytes and the proliferation of the capillaries into the mesenchyme marks an important step toward the fetus being able to survive outside the uterus. By the end of this stage, structural development has progressed sufficiently such that gas exchange is possible and neonatal survival can occur. Amniotic fluid is required for the canalicular stage to occur. Fetal lung growth is stimulated in part by the distending force of lung fluid in the airways, which occurs during fetal breathing and is inhibited by anhydramnios.
Saccular Phase
The saccular phase encompasses the period from 26 weeks until term, when the last generation of airspaces of the bronchial tree evolve. During this stage, there is a decrease in interstitial tissue, and a thinning of the airspace, resulting in a smaller population of collagen and elastic fibers. Surfactant production begins in the lungs. At the end of each respiratory tract passage, smooth-walled sacculi form, coated with type I and type II pneumocytes. Lamellar bodies of the type II cells store surfactant, which is rich in phosphatidylinositol, a necessary component for alveolar stability. Neonatal lung stability correlates with the number of lamellar bodies present. In the absence of surfactant, the lung alveoli are prone to collapse.
Alveolar Phase
The last phase of lung development spans the period from approximately 32 weeks’ gestation into early childhood. In addition to further surfactant production, lung development during this period is characterized by growth of more bronchioles and alveoli. This allows the gas-exchange tissues of the lungs to expand and makes them capable of moving more air as the neonate grows. This final stage of lung development primarily occurs during postnatal life, building on the several million alveoli already present.
Understanding these processes provides important insight into the aberrations in development associated with pulmonary lesions. The failure of normal physical development of the lung at any phase can result in pathologic lesions and potential respiratory compromise.
Embryology
Although an intact pulmonary system is not required for intrauterine viability, the prenatal development of the respiratory system is integral to ex utero life and survival. To allow successful transition to neonatal life, fetal lungs must proceed through both structural and functional maturation processes, which occur from the early embryonic period and continue after birth. During structural development, there is branching of the airways and development of alveolar spaces, allowing gas exchange to occur once the first breath is taken at delivery. Functional development results in the creation of a surfactant system. This system is composed of phospholipids that decrease alveolar surface tension, inhibiting alveolar collapse during exhalation. The surfactant system develops in the third trimester and is typically mature at 36 weeks’ gestation. Birth prior to the development of the surfactant system or completed pulmonary development results in neonatal respiratory compromise.
The respiratory diverticulum (lung bud) appears as an outgrowth of the ventral wall of the foregut. The epithelium of the internal lining of the larynx, trachea, bronchi, and lungs is entirely of endodermal origin. The cartilaginous, smooth muscle, and connective tissues of the trachea and lungs are derived from splanchnic mesoderm that surrounds the tubular framework. The lung bud is in open communication with the foregut. The diverticulum develops in a craniocaudal direction, leading to development of the upper respiratory structures (nose and pharynx) prior to the lower structures. Two tracheoesophageal ridges separate the diverticulum from the foregut. The dorsal portion of the foregut divides into the esophagus and the ventral portion into the trachea and lung buds.
The pulmonary circulation is a highly specialized vascular network, connecting the interdependent heart and lungs ( Fig. 12-1 ). The vascular bed parallels the airways and links the arterial and venous poles of the heart. The pulmonary vasculature has been described as appearing de novo within the mesoderm ventral and lateral to the foregut endoderm, suggesting that this mesoderm pool gives rise to the future pulmonary vasculature as lung develops from the foregut. The lungs have two sets of lymphatic vessels, a superficial set located beneath the pleura and a deep set which follows the blood vessels and extends along the bronchi. Both sets end within the bronchial glands. Efferent lymphatic vessels travel up the trachea, ending at the left-sided thoracic duct or the right-sided lymphatic duct. The fetal lungs play a key role in amniotic fluid volume maintenance: 15 mL/kg of body weight in fluid is produced by the lungs and flows out via the trachea and mouth, circulating and contributing to the amniotic fluid that is swallowed.
The basic structure of the diaphragm is also established early in gestation. Initially, the septum transversum develops, lying caudal to the heart and rostral to the umbilicus, in a position that eventually divides the intraembryonic cavity into the pleuropericardial cavity and the peritoneal cavity. The diaphragm subsequently undergoes programmed muscularization; this represents the final stage in the formation of the basic foundation of the diaphragm and is completed by 14 weeks’ gestation.
Fetal lung development occurs in five stages: embryonic, pseudoglandular, canalicular, saccular, and alveolar ( Fig. 12-2 ). The timing of these phases is approximate. Transitions between stages are gradual, with overlap from one stage to the next.
Embryonic Phase
The embryonic phase of lung development takes place during the first 4 to 5 weeks of gestation. During this phase, the larynx, trachea, and lung bud form from the foregut. The embryonic phase of lung development begins at about day 28 with the formation of a groove in the ventral lower pharynx, the sulcus laryngotrachealis. A few days later, at about day 30, a bud forms from the lower part, creating the true lung primordium. Development progresses in the 8-week-old embryo as the lobar buds subdivide and form the bronchopulmonary segments. Early on, the asymmetry of the main bronchi is established, with the smaller bud on the left directed more laterally than the larger one on the right, which is parallel to the esophagus and directed more caudally. The embryonic phase proceeds with the unequal dividing of the endodermal branches followed by further division. At the end of the embryonic period, the five lobes of the lungs (three right and two left) are present.
Pseudoglandular Phase
The pseudoglandular stage takes place between the 7th and 16th weeks of embryonic development. Conducting airways are formed by progressive branching of the original lung buds into smaller and more numerous areas. Each bud eventually becomes an independent respiratory unit, served by a bronchiole surrounded by capillary vessels that will bring blood to the lungs for oxygen. During this stage, the first differentiation of lung epithelium occurs. By 13 weeks’ gestation, cilia appear in the proximal airways.
Canalicular Phase
Lasting until approximately 25 weeks’ gestational age, the canalicular phase is crucial for the development of the gas-exchanging portion of the lung. By 20 weeks, there is differentiation into type I pneumocytes, the primary structural cell of the alveolus. Capillaries grow in close proximity to the distal surface of the alveolar cells. Lamellar bodies develop in type II alveolar cells and are the site of surfactant storage prior to release into the alveoli. A sufficient differentiation of the type II pneumocytes into the type I pneumocytes and the proliferation of the capillaries into the mesenchyme marks an important step toward the fetus being able to survive outside the uterus. By the end of this stage, structural development has progressed sufficiently such that gas exchange is possible and neonatal survival can occur. Amniotic fluid is required for the canalicular stage to occur. Fetal lung growth is stimulated in part by the distending force of lung fluid in the airways, which occurs during fetal breathing and is inhibited by anhydramnios.
Saccular Phase
The saccular phase encompasses the period from 26 weeks until term, when the last generation of airspaces of the bronchial tree evolve. During this stage, there is a decrease in interstitial tissue, and a thinning of the airspace, resulting in a smaller population of collagen and elastic fibers. Surfactant production begins in the lungs. At the end of each respiratory tract passage, smooth-walled sacculi form, coated with type I and type II pneumocytes. Lamellar bodies of the type II cells store surfactant, which is rich in phosphatidylinositol, a necessary component for alveolar stability. Neonatal lung stability correlates with the number of lamellar bodies present. In the absence of surfactant, the lung alveoli are prone to collapse.
Alveolar Phase
The last phase of lung development spans the period from approximately 32 weeks’ gestation into early childhood. In addition to further surfactant production, lung development during this period is characterized by growth of more bronchioles and alveoli. This allows the gas-exchange tissues of the lungs to expand and makes them capable of moving more air as the neonate grows. This final stage of lung development primarily occurs during postnatal life, building on the several million alveoli already present.
Understanding these processes provides important insight into the aberrations in development associated with pulmonary lesions. The failure of normal physical development of the lung at any phase can result in pathologic lesions and potential respiratory compromise.
Key Features for Assessment
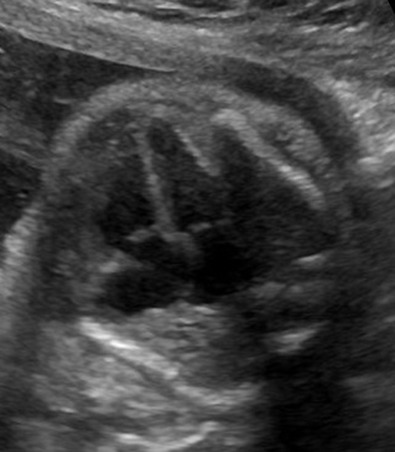
Complete assessment of the thoracic structures includes evaluation of lung structure and appearance. The lung fields should appear homogeneous with no fluid collections present ( Fig. 12-3 ). The size of the thorax relative to the fetal abdomen and the overall chest size relative to the size of the heart structures are nonspecific but are important components of evaluation. The cardiac circumference should be approximately 50% of the thoracic circumference. When the heart is large relative to thoracic size, further evaluation will determine if the chest is too small (potential pulmonary hypoplasia) or whether cardiomegaly is present. Abnormal cardiac axis or abnormal position within the chest may be a marker for displacement by a chest mass or a missing portion of the lung. Any abnormal cardiac position should prompt thorough evaluation of the lung fields. In addition to the homogeneous appearance of lung fields, the echogenicity of the lungs should also be assessed, with normal echogenicity being slightly greater than that of the fetal liver.
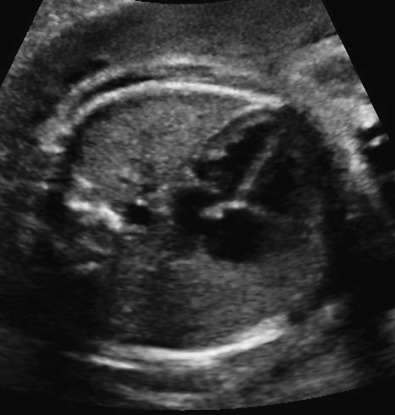
Adequate evaluation of the diaphragm requires sweeping side to side to assess continuity of the structure. The diaphragm will appear echolucent on ultrasound images ( Fig. 12-4 ). Shadowing from the ribs may present a challenge to visualization of the entire diaphragm. Particular care should be taken to examine the posterolateral portions, where most defects will occur. It is important that diaphragm continuity is confirmed and not just assumed based on other organs that are not appreciated in the chest. The contour of the diaphragm should also be assessed, as some abnormalities may be associated with eversion of the diaphragm downward.
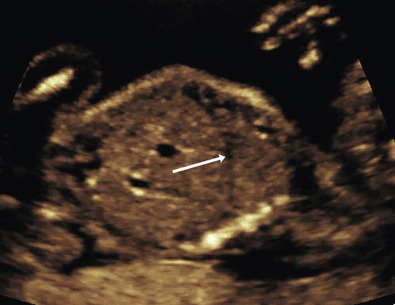
Pulmonary Hypoplasia
Pulmonary hypoplasia occurs as a spectrum characterized by incomplete development of unilateral or bilateral lung tissue. Clinically, it presents as respiratory compromise, with the severity reflecting the degree of reduction in lung size or in the number of lung cells, alveoli, or degree of bronchial branching. The severity is related to the timing of the insult in relation to the stage of embryologic development. Normal lung development requires adequate thoracic space as well as sufficient distention of the lungs through fluid exchange. Hypoplasia may therefore be caused by restriction or compression of developing fetal lung as well as by lack of amniotic fluid. Space-occupying lesions within the hemithorax, such as CDH, CPAM, massive cardiomegaly, and pleural effusion, can all cause pulmonary hypoplasia through mass effect, inhibiting normal formation of the pulmonary airways and alveoli. Thoracic musculoskeletal abnormalities, such as skeletal dysplasias and neuromuscular disorders, prohibit full expansion of the thoracic cage, with the restriction of chest size leading to hypoplasia. Additionally, oligohydramnios associated with renal or urinary tract abnormalities or preterm premature rupture of membranes during crucial embryologic stages reduces the development of normal pulmonary components. The mechanism by which lack of amniotic fluid inhibits lung growth is unknown. However, inhibition of fetal breathing movements or increased loss of fetal lung fluid into the amniotic space have been suggested as two possible mechanisms for pulmonary hypoplasia in the setting of oligohydramnios. The incidence of neonatal pulmonary hypoplasia with premature rupture of membranes between 18 and 26 weeks’ gestation ranges from 9% to 28%.
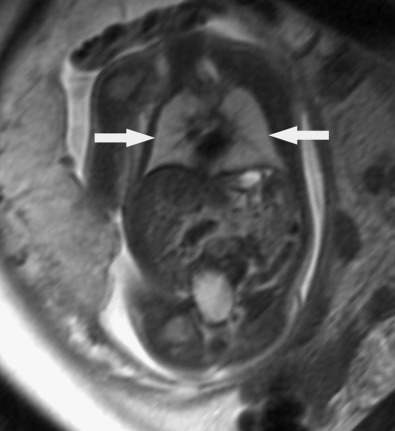
Although it is relatively uncommon, the high mortality rate associated with pulmonary hypoplasia makes prenatal prediction critical for appropriate counseling. Subjective ultrasound assessment of relative chest size is helpful only in extreme cases ( Fig. 12-5 ). Two-dimensional ultrasound measurements of fetal thoracic circumference, thoracic length, and the thoracic circumference to abdominal circumference (TC/AC) ratio have all been established ( Table 12-1 ) but also have limited predictive value except at the extremes. The use of the lung-to-head ratio (LHR) and lung volume measurements has been described more frequently with CDH (see later discussion under “ Congenital Diaphragmatic Hernia ”). Other methods, such as the quantitative lung index (QLI), have been employed in attempts to mathematically quantify lung volumes. This index, which is obtained by calculating lung area/(head circumference/10) , has been described as a calculation that is fetal age independent. It has been reported to have minimal variation throughout pregnancy, with near constant parameters. Recent studies have provided reference ranges for normal 20- to 36-week fetal lung areas and LHRs (using the longest diameter and area tracing methods). Unfortunately, these indirect measurements of lung volumes do not reliably and uniformly predict pulmonary hypoplasia, lung function, or likelihood of survival. The use of three-dimensional ultrasound to obtain volume measurements either directly or through the VOCAL technique (Virtual Organ Computer-Aided Analysis, General Electric Medical Systems) would seem more promising but have not been shown to be superior. MRI has also been used to calculate lung volumes ( Fig. 12-6 ). The risk for pulmonary hypoplasia is assessed by comparing lung volumes measured by MRI with normal reference values based on the gestational age. MRI has the advantage of more clearly delineating lung tissue from mediastinal structures and is not compromised by some of the limitations of ultrasound in the presence of oligohydramnios or high maternal body mass index (BMI). However, the ability of MRI to predict prognosis and survival has also been inconsistent, and the superiority of MRI in this regard remains controversial.
| Gestational Age (Week) | No. | PREDICTIVE PERCENTILES | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2.5 | 5 | 10 | 25 | 50 | 75 | 90 | 95 | 97.5 | ||
| 16 | 6 | 5.9 | 6.4 | 7.0 | 8.0 | 9.1 | 10.3 | 11.3 | 11.9 | 12.4 |
| 17 | 22 | 6.8 | 7.3 | 7.9 | 8.9 | 10.0 | 11.2 | 12.2 | 12.8 | 13.3 |
| 18 | 31 | 7.7 | 8.2 | 8.8 | 9.8 | 11.0 | 12.1 | 13.1 | 13.7 | 14.2 |
| 19 | 21 | 8.6 | 9.1 | 9.7 | 10.7 | 11.9 | 13.0 | 14.0 | 14.6 | 15.1 |
| 20 | 20 | 9.5 | 10.0 | 10.6 | 11.7 | 12.8 | 13.9 | 15.0 | 15.5 | 16.0 |
| 21 | 30 | 10.4 | 11.0 | 11.6 | 12.6 | 13.7 | 14.8 | 15.8 | 16.4 | 16.9 |
| 22 | 18 | 11.3 | 11.9 | 12.5 | 13.5 | 14.6 | 15.7 | 16.7 | 17.3 | 17.8 |
| 23 | 21 | 12.2 | 12.8 | 13.4 | 14.4 | 15.5 | 16.6 | 17.6 | 18.2 | 18.8 |
| 24 | 27 | 13.2 | 13.7 | 14.3 | 15.3 | 16.4 | 17.5 | 18.5 | 19.1 | 19.7 |
| 25 | 20 | 14.1 | 14.6 | 15.2 | 16.2 | 17.3 | 18.4 | 19.4 | 20.0 | 20.6 |
| 26 | 25 | 15.0 | 15.5 | 16.1 | 17.1 | 18.2 | 19.3 | 20.3 | 21.0 | 21.5 |
| 27 | 24 | 15.9 | 16.4 | 17.0 | 18.0 | 19.1 | 20.2 | 21.3 | 21.9 | 22.4 |
| 28 | 24 | 16.8 | 17.3 | 17.9 | 18.9 | 20.0 | 21.2 | 22.2 | 22.8 | 23.3 |
| 29 | 24 | 17.7 | 18.2 | 18.8 | 19.8 | 21.0 | 22.1 | 23.1 | 23.7 | 24.2 |
| 30 | 27 | 18.6 | 19.1 | 19.7 | 20.7 | 21.9 | 23.0 | 24.0 | 24.6 | 25.1 |
| 31 | 24 | 19.5 | 20.0 | 20.6 | 21.6 | 22.8 | 23.9 | 24.9 | 25.5 | 26.0 |
| 32 | 28 | 20.4 | 20.9 | 21.5 | 22.6 | 23.7 | 24.8 | 25.8 | 26.4 | 26.9 |
| 33 | 27 | 21.3 | 21.8 | 22.5 | 23.5 | 24.6 | 25.7 | 26.7 | 27.3 | 27.8 |
| 34 | 25 | 22.2 | 22.8 | 23.4 | 24.4 | 25.5 | 26.6 | 27.6 | 28.2 | 28.7 |
| 35 | 20 | 23.1 | 23.7 | 24.3 | 25.3 | 26.4 | 27.5 | 28.5 | 29.1 | 29.6 |
| 36 | 23 | 24.0 | 24.6 | 25.2 | 26.2 | 27.3 | 28.4 | 29.4 | 30.0 | 30.6 |
| 37 | 22 | 24.9 | 25.5 | 26.1 | 27.1 | 28.2 | 29.3 | 30.3 | 30.9 | 31.5 |
| 38 | 21 | 25.9 | 26.4 | 27.0 | 28.0 | 29.1 | 30.2 | 31.2 | 31.9 | 32.4 |
| 39 | 7 | 26.8 | 27.3 | 27.9 | 28.9 | 30.0 | 31.1 | 32.2 | 32.8 | 33.3 |
| 40 | 6 | 27.7 | 28.2 | 28.8 | 29.8 | 30.9 | 32.1 | 33.1 | 33.7 | 34.2 |
Pulmonary hypoplasia is associated with a need for prolonged respiratory support and can even result in infant death despite aggressive postnatal management. It is important to understand that neither sonography nor MRI uniformly predicts hypoplasia except in extreme cases. Despite the limitations, measurements obtained from both modalities may assist with predicted range of disease severity, allowing for appropriate counseling of families while sharing the limitations.
Congenital Pulmonary Airway Malformation
Fetal chest masses include CPAM (formerly referred to as congenital cystic adenomatoid malformation [CCAM]) and bronchopulmonary sequestration (BPS). CPAMs are the most common lung lesions identified by fetal ultrasound imaging. On occasion, hybrid lesions are found, with imaging and pathologic features of both CPAM and BPS. Historically, CPAMs were thought to be infrequent, with reported incidence between 1 in 25,000 and 1 in 35,000 live births. With ongoing improvements in ultrasound resolution, smaller CPAMs are now being diagnosed with increasing frequency, and the incidence of CPAM is now reported to be as high as 1 in 12,000 live births, accounting for 30% to 47% of fetal thoracic lung lesions. These lesions are most often unilateral (95%) and are rarely bilateral. The right and left lungs are involved with equal frequency, with the lower lobes more commonly affected. Current theories of pathogenesis suggest maturational arrest in bronchopulmonary development, resulting in dysplastic tissue distal to this segment.
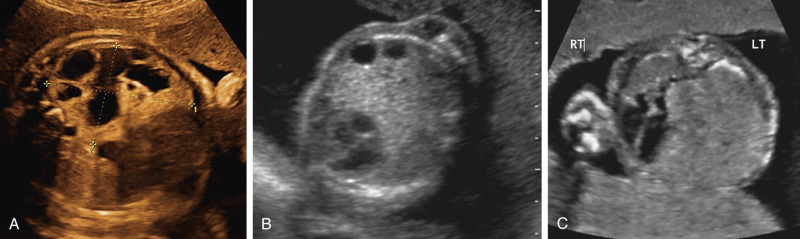
CPAMs have best been described as hamartomatous malformations of the lung characterized by abnormal branching of immature bronchioles. They communicate with the normal tracheobronchial tree and derive their blood supply from normal pulmonary circulation. These lesions develop during the pseudoglandular period, at 7 to 16 weeks. Stocker and associates initially classified the most common types of CPAMs into three categories. Type 1 is described as a lesion with a dominant cyst (3-10 cm in diameter); type 2 comprises multiple small cysts (0.5-2.0 cm diameter) that are distributed evenly and blend in with adjacent normal tissue; and type 3 is the small microcyst or what appears to be solid type of cyst on sonographic images ( Fig. 12-7 ). Recently, Stocker added two subtypes: type 0 (acinar dysplasia) and type 4 (large peripheral cyst of the distal acinus lined with alveolar cells), expanding the classification of CPAMs into five subtypes. However, this categorization was intended to be applied to resected lung specimens, rather than sonographic diagnoses. As the various types of CPAMs have little clinical significance, and the size and presence of larger cysts have more prognostic importance, the most common current practice is to describe the CPAM lesion as microcystic or macrocystic and to report its size.
Related posts:
 Ultrasound Evaluation of the Fetal Central Nervous System
Ultrasound Evaluation of the Fetal Central Nervous System
 Measurements Frequently Used to Estimate Gestational Age and Fetal Biometry
Measurements Frequently Used to Estimate Gestational Age and Fetal Biometry
 Role of Doppler Sonography in Obstetrics
Medications and Reported Associated Malformations
Role of Doppler Sonography in Obstetrics
Medications and Reported Associated Malformations
 Role of Sonography in Gynecologic Interventions
Role of Sonography in Gynecologic Interventions
 Ultrasound Features of Fetal Syndromes
Ultrasound Features of Fetal Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


