7 Urogenital Tract Definition: Incomplete closure of the bladder, lower urinary tract, symphysis, and lower abdominal wall. Incidence: One in 30 000 births. Sex ratio: M : F = 3 : 1. Clinical history/genetics: Mostly sporadic, rarely increased familial occurrence. Increase of alpha fetoprotein. If one parent is affected, there is a 1.5% risk of occurrence. Teratogens: Not known. Embryology: The two halves of the bladder fail to close in the midline, so that this is not considered to be primarily a defect of the abdominal wall, but rather a disturbed development of the urogenital sinus at around 7–9 weeks after menstruation. The urethra and clitoris or penis are also affected. Associated malformations: These are rarely found, except for the genital anomalies and clefting of the symphysis, which coexist with this malformation. Ultrasound findings: The urinary bladder cannot be detected on repeated scans. A lesion is seen in the lower part of the abdomen (eversion of the posterior bladder wall). The insertion of the umbilical cord is lower than normal. Clinical management: Further sonographic screening, including fetal echocardiography. Possibly karyotyping. Normal delivery. Procedure after birth: The defect should be covered with sterile cloth. Early surgical correction. Additional operative procedures are necessary in the first years of life. Prognosis: Surgical correction can restore urinary continence in 60–80% of cases. The survival rate is > 90%. Bladder cancers appear to occur more frequently in these patients. Self-Help Organization Title: Association for Bladder Exstrophy Community Description: Mutual support for persons affected by bladder exstrophy, including parents of children with bladder exstrophy, adults, health-care professionals and others interested in exstrophy. Newsletter, literature, information and referrals, informal pen-pal program, conferences, advocacy, directory of members. Informal kids’ e-mail exchange. Scope: International network Founded: 1991 Address: P.O. Box 1472, Wake Forest, NC 27588–1472, United States Telephone: 910–864–4308 E-mail: admin@bladderexstrophy.com References Cacciari A, Pilu GL, Mordenti M, Ceccarelli PL, Ruggeri G. Prenatal diagnosis of bladder exstrophy: what counseling? J Urol 1999; 161: 259–61. Gearhart JP, Ben Chaim J, Jeffs RD, Sanders RC. Criteria for the prenatal diagnosis of classic bladder exstrophy. Obstet Gynecol 1995; 85: 961–4. Goldstein I, Shalev E, Nisman D. The dilemma of prenatal diagnosis of bladder exstrophy: a case report and a review of the literature. Ultrasound Obstet Gynecol 2001; 17: 357–9. Langer JC, Brennan B, Lappalainen RE, et al. Cloacal exstrophy: prenatal diagnosis before rupture of the cloacal membrane. J Pediatr Surg 1992; 27: 1352–5. Mirk P, Calisti A, Fileni A. Prenatal sonographic diagnosis of bladder exstrophy. J Ultrasound Med 1986; 5: 291–3. Pinette MG, Pan YQ, Pinette SG, Stubblefield PG, Blastone J. Prenatal diagnosis of fetal bladder and cloacal exstrophy by ultrasound: a report of three cases. Reprod Med 1996; 41: 132–4. Thomas DF. Prenatal diagnosis: does it alter outcome? Prenat Diagn 2001; 21: 1004–11. Wilcox DT, Chitty LS. Non-visualisations of the fetal bladder: aetiology and management. Prenat Diagn 2001; 21: 977–83. Definition: In the prenatal scan, genital anomalies are diagnosed either as intersexual forms or as discrepancies between ultrasound findings and chromosomal findings. Clinical history/genetics: Heterogeneous causes are known: 1, chromosomal anomalies; 2, gene mutation; 3, combined chromosomal and molecular defects; 4, multifactorial inheritance; 5, nongenetic causes (Table 7.1). Incidence: With the exception of trisomy 13 and triploidy, chromosomal anomalies are rare. Single-gene anomalies such as the adrenogenital syndrome have an incidence of one in 15 000 births; hypospadias: one in 1000 male births. Associated malformations: Xp duplication: facial clefts, ventricular septal defect. In addition to pseudohermaphroditism and actual intersexuality, intersexual genitals are found in trisomy 13, triploidy, 13q syndrome, camptomelic dysplasia, Smith-Lemli–Opitz syndrome, and in other very rare syndromes. Ultrasound findings: Although the genitals are seen clearly, it is difficult to classify the gender correctly, the penis appearing too small or bent. The scrotal sac appears to be divided. A definite discrepancy is noted between the ultrasound sex determination and the chromosomal sex. Clinical management: If the family history suggests adrenogenital syndrome, chorionic villous sampling should be performed, and HLA typing is also possible. An affected female fetus can thus be treated very early in the prenatal stage (even at the end of the first trimester) to minimize virilization. Prognosis: This depends on the underlying disease; in adrenogenital syndrome, the prognosis is very favorable if therapy is started early. Self-Help Organization Title: Congenital Adrenal Hyperplasia Description: Offers educational and emotional support to families of children with congenital adrenal hyperplasia. Provides information and referrals, pen-pal program, phone support, annual convention, networking. Quarterly newsletter. Assistance in starting new groups. Fig. 7.1 Normal male genitals. 33 weeks of gestation.
Bladder Exstrophy
Genital Anomalies
| Chromosomal aberrations | |
| Xp, 1 p, 4 p, 9 p duplication; 10q deletion; trisomy 13; 17q aberration; triploidy; gonosomal mosaic (e.g., 45, X/6, XY) | Xp duplication: rare chromosomal aberration, genital anomalies of Y, double Xp chromosome: normal female genitalia or intersexual genitalia |
| Genetic mutations | |
| Adrenogenital syndrome (AGS) | Autosomal-recessive; the most common causes (in 70%) are mutations in the CYP21 gene for 21-hydroxylase, gene locus 6 p21 |
| Testicular feminization | Frequently X-recessive, gene mutation in the androgen receptor located at Xq11 – 12; any form between normal female genitals to intersexuality if 46,XY karyotype present |
| XY gonadal dysgenesis | Genetically heterogeneous; a frequent cause, in 10–15% of test persons, is a gene mutation in the testes-determining factor gene (SRY gene); a further 10–15% show deletion of the SRY gene, gene locus: Yp11.3. Depending on the expression of the gene mutation, there is either pure XY gonadal agenesis (Swyer syndrome) with normal female genitalia, or intersexual genitalia in incomplete gonadal dysgenesis. X-related recessive inheritance has been reported; autosomal-recessive inheritance with limited genital development is probable |
| Pure hermaphroditism | Possible combination of chromosomes: 46,XX, 46,XY or 46,XX/46,XY. Genetic cause largely unknown; SRY mutations have been seen in some cases, ovotestes or ovaries and testicles are present, external genitalia normal male or intersexual, presence of breasts |
| Smith-Lemli–Opitz syndrome, type II | Autosomal-recessive; hypospadias and cryptorchism if XY, no genital anomalies in female patients |
| Aarskog syndrome | X-related recessive, hypospadias |
| Genitopalatocardiac syndrome | Hypospadias or female genitalia in 46,XY karyotype, cardiac anomalies, cleft palate, club feet, kyphoscoliosis |
| Hypospadias | In rare cases, mutation in the SRY gene; mostly multifactorial inheritance (see below) |
| Denys-Drash syndrome | Gonadal dysgenesis; in 46,XY karyotype female or intersexual genitals, renal malformations, mutations in the Wilms tumor-1 gene (WT1) |
| Combined chromosomal aberration and molecular defect | |
| XX man | Normal female chromosomes. The gene for testes-determining factor (SRY gene) is incorrectly located on an X chromosome, a crossover that has failed in the germ-cell phase—this is the cause in 70% of XX men; the etiology for the rest is unclear. Normal male genitals are present |
| Camptomelic dysplasia | Two-thirds of patients have genital malformations with autosomaldominant inheritance |
| Multifactorial inheritance | |
| Hypospadias | If hypospadias is not associated with a syndrome, the recurrence rate for male siblings is 10% |
| Nongenetic causes | |
| Vanishing twin | Early intrauterine death of a twin, with residual cellular components. This may be responsible for discrepancies in genital findings (especially in CVS diagnosis) that cannot be explained by contamination by maternal cells |
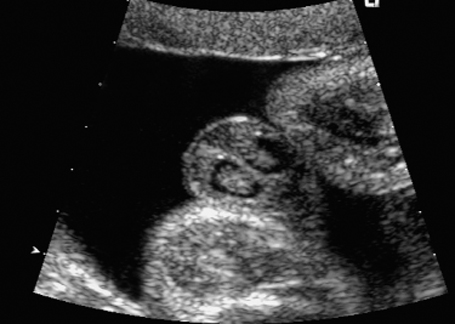
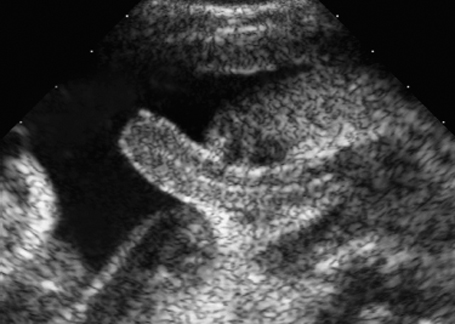
Fig. 7.2 Hydrocele. Fetal male gonads at 29 + 6 weeks. Bilateral hydroceles of the testes.
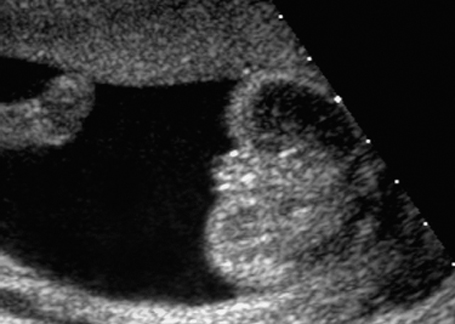
Fig. 7.3 Normal female genitals, 22 + 2 weeks. Cross-section showing labia majora and minora.
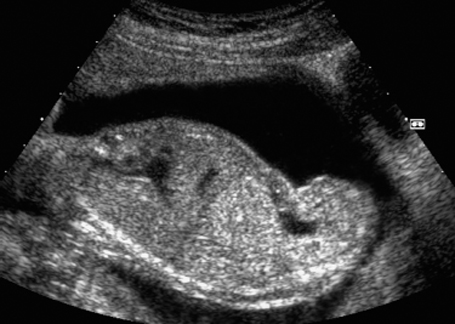
Fig. 7.4 Normal female genitals, 21 + 4 weeks. Longitudinal view showing the labia.
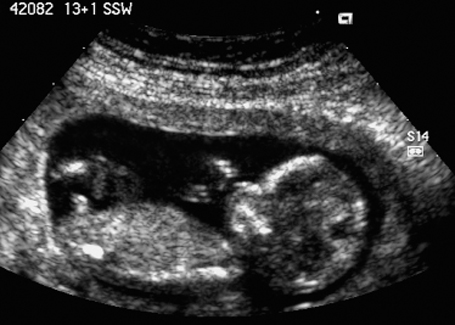
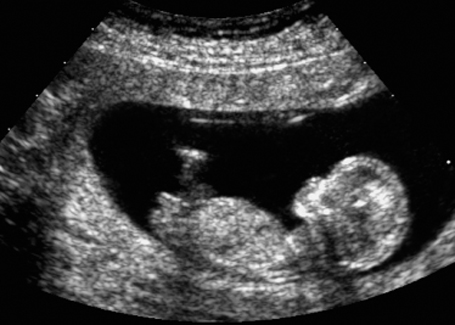
Fig. 7.6 Normal female genitals, 12 + 3 weeks. Longitudinal view. The clitoris is seen almost parallel to the body axis.
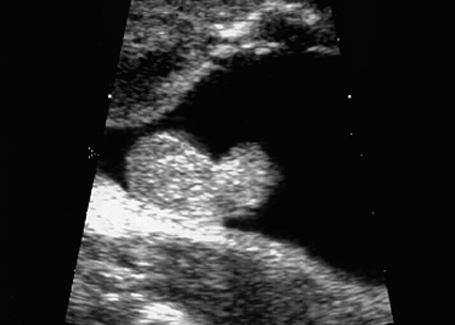
Fig. 7.7 Scrotal hypospadias. Genitals of a male fetus, longitudinal section. Atypical size and form of the penis: scrotal hypospadias, at 28 + 5 weeks.
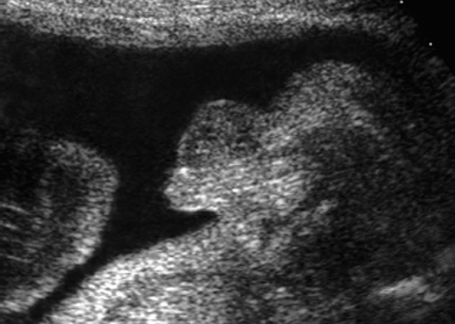
Fig. 7.8 Micropenis. Male genitals, longitudinal section. Micropenis, 28 + 4 weeks.
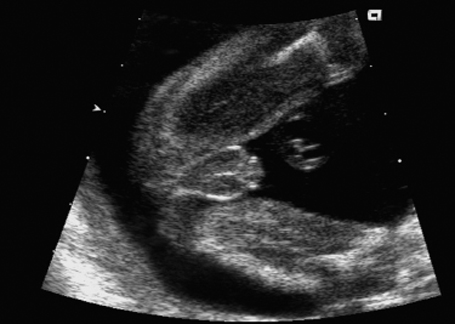
Fig. 7.9 Micropenis. Male genitals 22 + 1 weeks, frontal view.
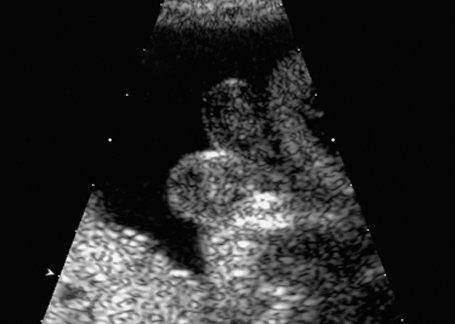
Fig. 7.10 Scrotum bipartitum, at 30 + 2 weeks, in a fetus with anal atresia.
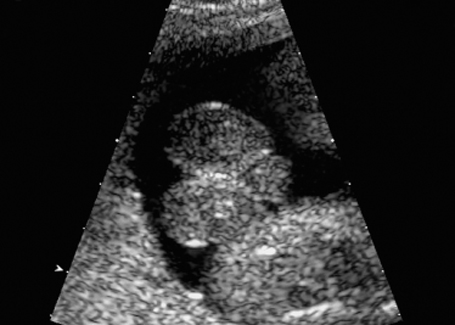
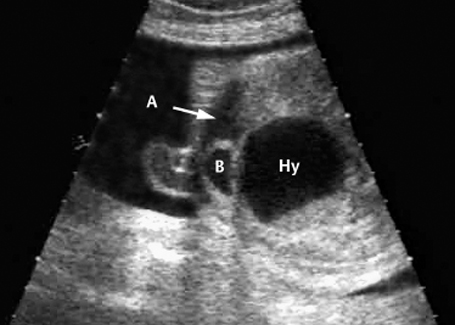
Fig. 7.12 Hydrometrocolpos in McKusick-Kaufmann syndrome, 32 + 3 weeks. A: ascites, B: bladder, Hy: hydrometrocolpos.
Scope: National network of MAGIC
Founded: 1993
Address: CAH Division of MAGIC Foundation, 1327 N. Harlem Ave., Oak Park, IL 60302, United States
Telephone: 1–800–362–4423
Fax: 708–383–0899
E-mail: mary@magicfoundation.org
References
Angle B, Tint GS, Yacoub OA, Clark AL. Atypical case of Smith-Lemli-Opitz syndrome: implications for diagnosis. Am J Med Genet 1998; 80: 322–6.
Cerame BI, Newfield RS, Pascoe L, et al. Prenatal diagnosis and treatment of 11-beta-hydroxylase deficiency congenital adrenal hyperplasia resulting in normal female genitalia. J Clin Endocrinol Metab 1999; 84: 3129–34.
Cheikhelard A, Luton D, Philippe-Chomette P, et al. How accurate is the prenatal diagnosis of abnormal genitalia? J Urol 2000; 164: 984–7.
Kolon TF, Gray CL, Borboroglu PG. Prenatal karyotype and ultrasound discordance in intersex conditions. Urology 1999; 54: 1097.
Kratz LE, Kelley RI. Prenatal diagnosis of the RSH/Smith–Lemli–Opitz syndrome. Am J Med Genet 1998; 376–81.
Meizner I, Mashiach R, Shalev J, Efrat Z, Feldberg D. The “tulip sign”: a sonographic clue for in-utero diagnosis of severe hypospadias. Ultrasound Obstet Gynecol 2002; 19: 250–3.
Neri G, Opitz J. Syndromal (and nonsyndromal) forms of male pseudohermaphroditism. Am J Med 1999: 89: 201–9.
Smith DP, Felker RE, Noe HN, Emerson DS, Mercer B. Prenatal diagnosis of genital anomalies. Urology 47: 114–7.
Speiser PW. Prenatal treatment of congenital adrenal hyperplasia. J Urol 1999; 162: 534–6.
Hydronephrosis
Definition: Dilation of the renal pelvis due to ureteropelvic, ureterovesical, vesicourethral stenosis or reflux. Hydronephrosis constitutes 75% of the renal anomalies detected prenatally. In 75% of cases, the finding is unilateral.
Incidence: One in 200 to 1 : 1000 births.
Sex ratio: M : F = 4 : 1 (ureteropelvic stenosis), M > F (ureterovesical stenosis).
Clinical history/genetics: Mostly sporadic, but may occur in association with 70 different syndromes.
Teratogens: Thalidomide, diabetes mellitus, cocaine, benzodiazepine.
Embryology: There are three different forms of obstruction—intrinsic, extrinsic, and secondary. The most common cause is the intrinsic form, due to an abnormal bundle of muscle leading to obstruction of the ureter. Over a certain period of time, this bundle degenerates, and the resulting fibrous tissue causes compression of the ureter. In the extrinsic form, the compression is caused primarily by a lesion outside of the ureter, usually a blood vessel anomaly (an accessory renalartery). The secondary form is due to ureterovesical reflux mimicking urinary tract obstruction.
Associated malformations: In 30% of cases, bilateral ureteropelvic stenoses are found. Other urogenital anomalies are found in a further 30%: cloaca formation, bladder exstrophy, ureterocele, megacystis, megaureter, urethral valve syndrome, ureter obstruction, vesicoureteral reflux. In addition, intra-abdominal tumors and cardiac anomalies may also be found.
Associated syndromes: In 20% of cases, multiple anomalies or syndromes coexist; chromosomal anomalies are found in 5%.
Ultrasound findings: Widening of the renal pelvis is well detected during abdominal scanning. The width of the renal pelvis should be measured in its anteroposterior diameter in a cross-sectional scan. The transverse or oblique measurements give higher values, which are incorrect. It is difficult to detect widening of the ureter. In severe cases, the ureter can be followed up to its entry into the bladder. The upper limits for the widening of the renal pelvis are 5 mm around 20 weeks, and up to 10 mm after 30 weeks. The presence of oligohydramnios is an unfavorable sign.
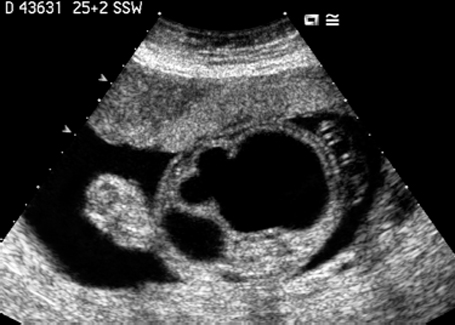
Fig. 7.13 Hydronephrosis. Severe bilateral hydronephrosis at 25 + 2 weeks. The urinary bladder appears normal, and there is a normal amount of amniotic fluid.
Procedure after birth: An ultrasound examination should be carried out on the second or the third day of life. Immediately after birth, diuresis is usually compromised due to the stress of labor, and renal congestion is not as pronounced as it is after a couple of days. Many cases do not require surgical correction. In these cases, an ultrasound check-up is recommended at 4 weeks. If reflux is diagnosed, antibiotics can be given prophylactically to prevent infection. Which surgical measures are taken (such as pyeloplasty or nephrostomy for decompression of obstruction over several months) depends on the severity of the findings and on the urologist’s opinion.
Prognosis: After birth, spontaneous remission is seen in 40% of the cases diagnosed at the prenatal stage. In the first years of life, treatment-resistant reflux may cause irreversible renal damage due to recurrent urinary tract infection. Surgical corrections are very successful.
References
Bobrowski RA, Levin RB, Lauria MR, Treadwell MC, Gonik B, Bottoms SF. In utero progression of isolated renal pelvis dilation. Am J Perinatol 1997; 14: 423–6.
Chertin B, Rolle U, Farkas A, Puri P. Does delaying pyelo-plasty affect renal function in children with a prenatal diagnosis of pelvi-ureteric junction obstruction? BJU Int 2002; 90: 72–5.
Cooper CS, Andrews JI, Hansen WF, Yankowitz J. Antenatal hydronephrosis: evaluation and outcome. Curr Urol Rep 2002; 3: 131–8.
Coplen DE. Prenatal intervention for hydronephrosis. J Urol 1997; 157: 2270–7.
Fugelseth D, Lindemann R, Sande HA, Refsum S, Nordshus T. Prenatal diagnosis of urinary tract anomalies; the value of two ultrasound examinations. Acta Obstet Gynecol Scand 1994; 73: 290–3.
Meizner I, Yitzhak M, Levi A, Barki Y, Barnhard Y, Glezerman M. Fetal pelvic kidney: a challenge in prenatal diagnosis? Ultrasound Obstet Gynecol 1995; 5: 391–3.
Nonomura K, Yamashita T, Kanagawa K, Itoh K, Koyanagi T. Management and outcome of antenatally diagnosed hydronephrosis. Int J Urol 1994; 1: 121–8.
Onen A, Jayanthi VR, Koff SA. Long-term follow-up of prenatally detected severe bilateral newborn hydronephrosis initially managed nonoperatively. J Urol 2002; 168: 1118–20.
Peters CA. The long-term follow-up of prenatally detected severe bilateral newborn hydronephrosis initially managed nonoperatively [editorial]. J Urol 2002; 168: 1121–2.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


