1 Brain and Extra-axial Lesions(Table 1.7 – Table 1.8)
Lesions | CT Findings | Comments |
Neoplasms | ||
Astrocytoma | Low-grade astrocytoma: Focal or diffuse mass lesion with low to intermediate attenuation, with or without mild contrast enhancement. Minimal associated mass effect. Juvenile pilocytic astrocytoma, subtype: Solid/cystic focal lesion with low to intermediate attenuation signal, usually with contrast enhancement. Lesions located in the cerebellum, hypothalamus, adjacent to third or fourth ventricles, brainstem. | Often occurs in children and adults (20–40 y). Tumors comprised of well-differentiated astrocytes. Association with neurofibromatosis type 1; 10-y survival; may become malignant. Common in children, usually favorable prognosis if totally resected. |
| Gliomatosis cerebri: Infiltrative lesion with poorly defined margins with mass effect located in the white matter that can extend into the basal ganglia, with low to intermediate attenuation, usually no contrast enhancement until late in disease. | Diffusely infiltrating astrocytoma with relative preservation of underlying brain architecture. Imaging appearance may be more prognostic than histologic grade; ~2 y survival. |
| Anaplastic astrocytoma: Often irregularly marginated lesion located in the white matter and extending into the basal ganglia with low to intermediate attenuation, with or without contrast enhancement. | Intermediate between low-grade astrocytoma and glioblastoma multiforme; ~2- y survival. |
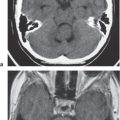
Glioblastoma multiforme | Irregularly marginated mass lesion with necrosis or cyst, mixed attenuation images, heterogeneous, with or without hemorrhage, prominent heterogeneous contrast enhancement; peripheral edema; can cross the corpus callosum. | Most common primary CNS tumor, highly malignant neoplasms with necrosis and vascular proliferation; usually seen in patients older than 50 y; extent of lesion underestimated by CT and MRI; survival < 1 y. |
Giant cell astrocytoma/tuberous sclerosis | Circumscribed lesion located near the foramen of Monro with mixed low to intermediate attenuation with or without cysts and/or calcifications, with heterogeneous contrast enhancement. | Subependymal hamartoma near the foramen of Monro; occurs in 15% of patients with tuberous sclerosis < 20 y; slow-growing lesions can progressively cause obstruction of CSF flow through the foramen of Monro. Long-term survival usual if resected. |
Hamartoma/tuberous sclerosis | Cortical-subcortical lesion with variable attenuation; calcifications in 50% of older children; contrast enhancement uncommon. Subependymal hamartomas: Small nodules located along and projecting into the lateral ventricles; calcification and contrast enhancement common. | Cortical and subependymal hamartomas are nonmalignant lesions associated with tuberous sclerosis. Tuberous sclerosis is an autosomal dominant disorder associated with hamartomas in multiple organs. |
Pleomorphic xanthoastrocytoma | Circumscribed supratentorial lesion; low to intermediate attenuation, heterogeneous contrast enhancement, with or without enhancing mural nodule associated with cyst. | Rare type of astrocytoma occurring in young adults and children; associated with seizure history. |
Oligodendroglioma | Circumscribed lesion with mixed low to intermediate attenuation; with or without areas of clumplike calcification, heterogeneous contrast enhancement. | Uncommon slow-growing gliomas with usually mixed histologic patterns (e.g., astrocytomas). Usually in adults older than 35 y; 85% supratentorial. If low grade, 75% 5-y survival; higher grade lesions have a worse prognosis. |
Ganglioglioma, ganglioneuroma, gangliocytoma | Circumscribed tumor, usually supratentorial, often temporal or frontal lobes; low to intermediate attenuation, with or without cysts, with or without contrast enhancement. | Ganglioglioma (contains glial and neuronal elements), ganglioneuroma (contains only ganglion cells). Uncommon tumors, < 30 y, seizure presentation, slow-growing neoplasms. Gangliocytoma (contains only neuronal elements, dysplastic brain tissue). Favorable prognosis if completely resected. |
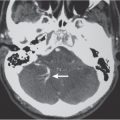
Ependymoma | Circumscribed lobulated supratentorial lesion, often extraventricular, with or without cysts and/or calcifications, variable contrast enhancement. | Occurs more commonly in children than adults; one third supratentorial, two thirds infratentorial; 45% 5-y survival. |
Lymphoma | Primary CNS lymphoma: Focal or infiltrating lesion located in the basal ganglia, periventricular regions, posterior fossa/brainstem; low to intermediate attenuation, with or without hemorrhage/necrosis in immunocompromised patients; usually show contrast enhancement. Diffuse leptomeningeal enhancement is another pattern of intracranial lymphoma. | Primary CNS lymphoma more common than secondary, usually in adults older than 40 y. B-cell lymphoma more common than T-cell lymphoma; increasing incidence related to number of immunocompromised patients in population. CT and MRI imaging features of primary and secondary lymphoma of brain overlap. Intracranial lymphoma can involve the leptomeninges in secondary lymphoma > primary lymphoma. |
Metastases | Circumscribed spheroid lesions in the brain, usually low to intermediate attenuation with or without hemorrhage, calcifications, cysts; variable contrast enhancement, often low attenuation peripheral to nodular-enhancing lesion representing axonal edema. | Represent ~33% of intracranial tumors, usually from extracranial primary neoplasm in adults older than 40 y. Primary tumor source: lung > breast > GI > GU > melanoma. |
Tumorlike lesions | ||
Neuroepithelial cyst | Well-circumscribed cysts with low attenuation similar to CSF; thin walls; no contrast enhancement or peripheral edema. | Cyst walls have histopathologic features similar to epithelium; neuroepithelial cysts located in choroid plexus > choroidal fissure > ventricles > brain parenchyma. |
Perivascular spaces | Focus or foci with attenuation similar to CSF, no contrast enhancement; located in the basal ganglia, high subcortical white matter/centrum semiovale. | Pial-lined spaces filled with CSF containing arteries supplying brain parenchyma; also referred to as Virchow-Robin spaces. Perivascular spaces increase in size and number with aging. |
Infections | ||
Cerebritis | Poorly defined zone or focal area of low to intermediate attenuation signal; minimal or no contrast enhancement; involves cerebral cortex and white matter for bacterial and fungal infections. | Focal infection/inflammation of brain tissue from bacteria or fungi; secondary to sinusitis, meningitis, surgery, hematogenous source (cardiac and other vascular shunts), and/or immunocompromised status. Can progress to abscess formation. |
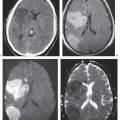
Pyogenic brain abscess | Circumscribed lesion with low attenuation signal (with or without air-fluid level) surrounded by a thin rim of low to intermediate attenuation that shows contrast enhancement surrounded by a peripheral poorly defined zone of low attenuation representing edema; ringlike zone of contrast enhancement is sometimes thicker laterally than medially. | Formation of brain abscess occurs 2 weeks after cerebritis with liquefaction and necrosis centrally surrounded by a capsule and peripheral edema. Can be multiple. Complication from meningitis and/or sinusitis, septicemia, trauma, surgery, and cardiac shunt. |
Fungal brain infection/Abscess | Vary depending on organism; lesions occur in meninges and brain parenchyma, solid or cystic lesions with low to intermediate attenuation, nodular or ring enhancement, peripheral low attenuation in brain representing edema. | Occur in immunocompromised or diabetic patients with resultant granulomas in meninges and brain parenchyma. Cryptococcus involves the basal meninges and extends along perivascular spaces into the basal ganglia; Aspergillus and Mucor spread via direct extension through paranasal sinuses or hematogenously, invade blood vessels resulting in hemorrhagic lesions and/or cerebral infarcts; coccidioidomycosis usually involves the basal meninges. |
Tuberculoma | Intra-axial lesions in cerebral hemispheres and basal ganglia (adults) and cerebellum (children): low to intermediate attenuation; with solid or rim pattern of contrast enhancement; with or without calcification. Meningeal lesions: Nodular or cystic-appearing zones of basilar meningeal contrast enhancement. | Occurs in immunocompromised patients and in inhabitants in developing countries. Caseating intracranial granulomas via hematogenous dissemination; meninges > brain lesions. |
Encephalitis | Poorly defined zone of low to intermediate attenuation. Minimal or no contrast enhancement; involves cerebral cortex and/or white matter; minimal localized mass effect. Herpes simplex typically involves the temporal lobes/limbic system with or without hemorrhage; CMV usually in periventricular location. | Infection/inflammation of brain tissue from viruses, often in immunocompromised patients (e.g., herpes simplex, CMV, and progressive multifocal leukoencephalopathy) or immunocompetent patients (e.g., St. Louis encephalitis, eastern or western equine encephalitis, and Epstein-Barr virus). |
Rasmussen encephalitis | Progressive atrophy of one cerebral hemisphere with poorly defined zones of low attenuation involving the white matter, basal ganglia, and cortex; usually no contrast enhancement. | Usually seen in children younger than 10 y; severe and progressive epilepsy and unilateral neurologic deficits (hemiplegia, psychomotor deterioration, chronic slow viral infectious process possibly caused by CMV or Epstein-Barr virus). Treatment: hemispherectomy. |
Creutzfeldt-Jakob disease | Progressive cerebral atrophy. Typically no contrast enhancement. | Spongiform encephalopathy caused by slow infection from prion (proteinaceous infectious particle); usually seen in adults 40 to 80 y, progressive dementia, <10% survive more than 2 y. |
Parasitic brain lesions | ||
Toxoplasmosis | Single or multiple solid and/or cystic-appearing lesions located in basal ganglia and/or corticomedullary junctions in cerebral hemispheres, low to intermediate attenuation; nodular or rim pattern of contrast enhancement, with or without peripheral low attenuation (edema). | Most common opportunistic CNS infection in AIDS patients; caused by ingestion of food contaminated with parasites (Toxoplasma gondii). |
Cysticercosis | Single or multiple cystic-appearing lesions in brain or meninges. Acute/subacute phase: Low to intermediate attenuation, with or without rim and/or nodular pattern of contrast enhancement, with or without peripheral low attenuation (edema). Chronic phase: Calcified granulomas. | Caused by ingestion of ova (Taenia solium) in contaminated food (undercooked pork); involves meninges > brain parenchyma > ventricles. |
Hydatid cyst | Echinococcus granulosus: Single or rarely multiple cystic lesions with low attenuation with a thin wall with intermediate attenuation; typically no contrast enhancement or peripheral edema unless super-infected; often located in vascular territory of the middle cerebral artery. Echinococcus multilocularis: Cystic (with or without multilocular) and/or solid lesions; central zone of low attenuation surrounded by a slightly thickened rim of intermediate attenuation, with contrast enhancement; peripheral zone of low attenuation (edema) and calcifications are common. | Caused by parasites E. granulosus (South America, Middle East, Australia, and New Zealand) and E. multilocularis (North America, Europe, Turkey, and China). CNS involvement in 2% of cases of hydatid infestation. |
Inflammatory lesions | ||
Demyelinating Disease: multiple sclerosis, ADEM | Lesions located in cerebral or cerebellar white matter, brainstem, and basal ganglia; lesions usually have low to intermediate attenuation. With or without contrast enhancement. Contrast enhancement can be ringlike or nodular, usually in acute/early subacute phase of demyelination. Lesions rarely can have associated mass effect simulating neoplasms. | Multiple sclerosis is the most common acquired demyelinating disease usually affecting women (peak ages 20–40 y). Other demyelinating diseases are acute disseminated encephalomyelitis/immune mediated demyelination after viral infection; toxins (exogenous from environmental exposure or ingestion of alcohol, solvents, etc.) or endogenous from metabolic disorder (leukodystrophies, mitochondrial encephalopathies, etc.), radiation injury, trauma, or vascular disease. |
Sarcoidosis | Poorly marginated intra-axial zone with low to intermediate attenuation, usually shows contrast enhancement, with localized mass effect and peripheral edema. Often associated with contrast enhancement in the leptomeninges. | Multisystem noncaseating granulomatous disease of uncertain cause that can involve the CNS in 5% to 15% of cases. Associated with severe neurologic deficits if untreated. |
Radiation Necrosis | Focal lesion with or without mass effect or poorly defined zone of low to intermediate attenuation, with or without contrast enhancement involving tissue (gray matter and/or white matter) in field of treatment. | Usually occurs from 4 to 6 months to 10 y after radiation treatment; may be difficult to distinguish from neoplasm. PET and magnetic resonance hydrogen spectroscopy might be helpful for evaluation. |
Vascular lesions | ||
Cerebral infarction | Ischemic anoxic or lacunar infarcts: CT features of cerebral infarcts depend on age of infarct relative to time of examination. Hyperacute phase (< 12 h): CT: No abnormality (50%), decreased attenuation and blurring of lentiform nuclei; hyperdense artery in up to 50% of cases. MRI: Localized edema, usually isointense signal to normal brain on T1- and T2-weighted images. Diffusion-weighted images can show positive findings related to decreased apparent diffusion coefficients secondary to cytotoxic edema, absence of arterial flow void, or arterial enhancement in the vascular distribution of the infarct. Acute (12–24 h): CT: Zones with decreased attenuation in basal ganglia, blurring of junction between cerebral cortex and white matter, sulcal effacement. MRI: Intermediate signal on T1-weighted images, high signal on T2-weighted images, localized edema. Signal abnormalities commonly involve the basal ganglia. Early subacute phase (24 h–3 d): CT: Localized swelling at sites with low attenuation involving gray and white matter (often wedge shaped), with or without hemorrhage. MRI: Zones with low to intermediate signal on T1-weighted images, high signal on T2-weighted images, localized edema, with or without hemorrhage, with or without gadolinium contrast enhancement. Late subacute phase (4 d–2 wk): CT: Localized swelling increases and then decreases; low attenuation at lesion can become more prominent, with or without gyral contrast enhancement. MRI: Low to intermediate signal on T1-weighted images, high signal on T2-weighted images; edema/mass effect diminishing, with or without hemorrhage, with or without enhancement. 2 weeks to 2 months: CT: With or without gyral contrast enhancement; localized mass effect resolves. MRI: Low to intermediate signal on T1-weighted images, high signal on T2-weighted images; edema resolves; with or without hemorrhage, with or without enhancement; eventually declines. > 2 months: CT: Zone of low attenuation associated with encephalomalacia. MRI: Low signal on T1-weighted images, high signal on T2-weighted images; encephalomalacic changes; with or without calcification, hemosiderin. | Cerebral infarcts usually result from occlusive vascular disease involving large, medium, or small arteries. Vascular occlusion may be secondary to atheromatous arterial disease, hypertension, cardiogenic emboli, neoplastic encasement, hypercoagulable states, dissection, congenital anomalies or inherited disorders, such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Cerebral infarcts usually result from arterial occlusion involving specific vascular territories, although occasionally they occur from metabolic disorders (mitochondrial encephalopathies, etc.) or intracranial venous occlusion (thrombophlebitis, hypercoagulable states, dehydration, etc., which do not correspond to arterial distributions). |
Hypoxic ischemic encephalopathy | Bilateral zones of low attenuation in the basal ganglia, caudate nuclei, thalami, brainstem, and/or perisylvian cerebral cortex. | Prolonged hypotension and anoxia results in ischemia and infarction in portions of the brain with selective vulnerability to hypoxia and impairment of aerobic metabolism. Can result from drowning, asphyxiation, or cardiac arrest. |
AVM | Lesions with irregular margins that can be located in the brain parenchyma (pia, dura, or both locations). AVMs contain multiple tortuous tubular vessels consisting of patent arteries with high blood flow, as well as thrombosed vessels, areas of hemorrhage in various phases, calcifications, and gliosis. The patent arterial and venous portions often show contrast enhancement on CTAs. CTAs can provide additional detailed information about the nidus, feeding arteries, and draining veins, as well as the presence of associated aneurysms. Usually not associated with mass effect unless there is recent hemorrhage or venous occlusion. | Infratentorial AVMs much less common than supratentorial AVMs. |
Cavernous hemangioma | Single or multiple multilobulated intra-axial lesions. CT: Lesions have intermediate to slightly increased attenuation, with or without calcifications, variable contrast enhancement (most show no or minimal enhancement, few show more prominent enhancement). MRI: Peripheral rim or irregular zone of low signal on T2-weighted images secondary to hemosiderin, surrounding a central zone of variable signal (low, intermediate, high, or mixed) on T1- and T2-weighted images depending on ages of hemorrhagic portions. Gradient echo techniques useful for detecting multiple lesions. | Intra-axial lesions composed of endothelial sinusoidal vessels under low pressure. Can be located in many different locations; multiple lesions > 50%. Association with venous angiomas and risk of hemorrhage causing seizures. |
Venous angioma | On postcontrast images, venous angiomas are seen as a contrast-enhancing transcortical vein draining a collection of small medullary veins (caput medusae). | Considered an anomalous venous formation typically not associated with hemorrhage; usually an incidental finding except when associated with cavernous hemangioma. |
Moyamoya | Multiple tortuous tubular vessels seen in the basal ganglia and thalami secondary to dilated collateral arteries, with contrast enhancement of these arteries related to slow flow within these collateral arteries versus normal-sized arteries. Often with contrast enhancement of the leptomeninges related to pial collateral vessels. Decreased or absent caliber of contrast-enhanced supraclinoid portions of the internal carotid arteries and proximal middle and anterior cerebral arteries. CTA shows stenosis and occlusion of the distal internal carotid arteries with collateral arteries (lenticulostriate, thalamoperforate, and leptomeningeal); best seen after contrast administration enabling detection of slow blood flow. | Progressive occlusive disease of the intracranial portions of the internal carotid arteries with resultant numerous dilated collateral arteries arising from the lenticulostriate and thalamoperforate arteries, as well as other parenchymal, leptomeningeal, and transdural arterial anastomoses. Term translated as “puffof smoke,” referring to the angiographic appearance of the collateral arteries (lenticulostriate, thalamoperforate). Usually nonspecific etiology but can be associated with neurofibromatosis, radiation-angiopathy, atherosclerosis, and sickle cell disease; usually children > adults in Asia. |
Hematoma | The attenuation of the hematoma depends on its age, size, location, hematocrit, hemoglobin oxidation state, clot retraction, and extent of edema. Hyperacute phase (4–6 h): Hemoglobin primarily as diamagnetic oxyhemoglobin (iron Fe2+ state). CT: High attenuation on CT. MRI: Intermediate signal on T1-weighted images, slightly high signal on T2-weighted images. Acute phase (12–48 h): Hemoglobin primarily as paramagnetic deoxyhemoglobin (iron, Fe2+ state). CT: High attenuation in acute clot directly related to hematocrit, hemoglobin concentration, and high protein concentration. Hematocrit in acute clot approaches 90%. MRI: Intermediate signal on T1-weighted images, low signal on T2-weighted images, surrounded by a peripheral zone of high T2 signal (edema). Early subacute phase (> 2 d): Hemoglobin becomes oxidized to the iron Fe3+ state, methemoglobin, which is strongly paramagnetic. CT: High attenuation. MRI: When methemoglobin is initially intracellular, the hematoma has high signal on T1-weighted images progressing from peripheral to central and low signal on T2-weighted images, surrounded by a zone of high T2 signal (edema). When methemoglobin eventually becomes primarily extracellular, the hematoma has high signal on T1-weighted images and high signal on T2-weighted images. Late subacute phase (> 7 d–6 wk): Intracerebral hematomas decrease 1.5 HU per day. Hematomas become isodense to hypodense; peripheral contrast enhancement from blood–brain barrier breakdown and vascularized capsule. Chronic phase: Hemoglobin as extracellular met-hemoglobin is progressively degraded to hemosiderin. CT: Chronic hematomas have low attenuation with localized encephalomalacia. Zone with high attenuation represents new sites of rebleeding. MRI: The hematoma progresses from a lesion with high signal on T1- and T2-weighted images with a peripheral rim of low signal on T2-weighted images (hemosiderin) to predominant hemosiderin composition with low signal on T2-weighted images. | Can result from trauma, ruptured aneurysms or vascular malformations, coagulopathy, hypertension, adverse drug reaction, amyloid angiopathy, hemorrhagic transformation of cerebral infarction, metastases, abscesses, and viral infections (herpes simplex, CMV). |
Toxic/metabolic abnormalities | ||
Carbon monoxide poisoning | CT: Acute poisoning shows symmetric decreased attenuation in the globus pallidi. MRI: Low signal on T1-weighted images and high signal on T2-weighted images in the putamen and globus pallidus bilaterally, with or without patchy contrast enhancement in necrotic zones. Diffusion-weighted images show restricted diffusion from acute necrosis. Similar but less pronounced findings are seen in the brainstem and cerebellum. | Toxic effects of CO result in selective necrosis of basal ganglia bilaterally and also to a lesser degree the brainstem and cerebellum. Atrophic changes involving the brain can be seen later that may be associated with cognitive impairment. The binding of CO to the heme protein is 250 times greater than oxygen, resulting in tissue hypoxia. |
Methanol intoxication | CT: Acute poisoning shows symmetric decreased attenuation in the basal ganglia and subcortical white matter, with or without hemorrhage. MRI: High signal on T2-weighted images in the putamen and globus pallidus bilaterally, with or without hemorrhage, with or without contrast enhancement. | Toxic effects result in selective necrosis of basal ganglia/putamina bilaterally and subcortical white matter 12 to 24 hours after ingestion secondary to metabolic conversion by hepatic alcohol dehydrogenase to the toxin formic acid. |
Mitochondrial encephalopathy, lactic acidosis, and strokelike events (MELAS) and myoclonic epilepsy, ragged red fiber (MERRF) syndromes | CT: Symmetric zones of low attenuation in the basal ganglia and cerebral infarction that is not limited to one vascular distribution. MRI: High T2 signal in basal ganglia usually symmetric, as well as high T2 signal in cerebral and cerebellar cortex and subcortical white matter not corresponding to a specific large arterial vascular territory. Signal abnormalities may resolve and reappear. | MELAS is a maternally inherited disease affecting transfer RNA in mitochondria. MERRF is a mitochondrial encephalopathy associated with muscle weakness and myoclonic epilepsy, short stature, ophthalmoplegia, and cardiac disease. |
Leigh disease | CT: Zones of low attenuation in both caudate nuclei and putamina, with or without decreased attenuation in white matter; typically no contrast enhancement. MRI: Symmetric high signal on T2-weighted images in the globus pallidus, putamen, and caudate, as well as high signal on T2-weighted images in the thalami, cerebral and cerebellar white matter, cerebellar cortex, brainstem, and spinal cord gray matter; typically no gadolinium contrast enhancement. | Autosomal recessive disorder, also referred to as subacute necrotizing encephalopathy, occurs in three forms (infantile, juvenile, and adult onset); etiology related to abnormalities in oxidative metabolism in mitochondria from one of several defective enzymes; progressive neurodegenerative disease. Lesions in brainstem are associated with loss of respiratory control. |
Kearns-Sayre syndrome | CT: Zones of low attenuation in both caudate nuclei and putamina, with or without decreased attenuation in white matter, with or without calcifications of basal ganglia, thalami, and dentate nuclei. MRI: Symmetric high signal on T2-weighted images in the globus pallidus, putamen, and caudate; high signal on T2-weighted images in the thalami, cerebral and cerebellar white matter, cerebellar cortex, and brainstem; typically no gadolinium enhancement. | Mitochondrial disorder associated with external ophthalmoplegia, retinitis pigmentosa, and onset of clinical muscular and neurologic signs in patients younger than 20 y. |
Cockayne syndrome | CT: Calcifications in basal ganglia and dentate nuclei, zones of low attenuation in cerebral white matter. MRI: High signal on T2-weighted images involving the periventricular white matter, basal ganglia, and dentate nuclei, with calcifications in basal ganglia and dentate nuclei, progressive cerebral and cerebellar atrophy, microcephaly. | Autosomal recessive disorder with deficient repair mechanisms for DNA; presents in first decade with progressive neurologic dysfunction, cataracts, cutaneous photosensitivity, optic atrophy, and dwarfism. |
Wilson disease | CT: Zones of low attenuation in the basal ganglia and thalami. No abnormal contrast enhancement seen. Progressive atrophy of the cerebrum, cerebellum, and brainstem. MRI: High signal on T2-weighted images in the putamen bilaterally, as well as in the thalami, caudate nuclei, dentate nuclei, and brainstem (periaqueductal zone). Low signal on T2-weighted images can also be seen in the caudate and putamen. Progressive atrophy of the cerebrum, cerebellum, and brainstem. | Autosomal recessive disease manifest by decreased functional serum ceruloplasmin levels and altered copper metabolism with increased urinary excretion of copper. Usually presents in childhood with abnormal toxic copper deposition in tissues, resulting in cirrhosis and degenerative changes in the basal ganglia (lentiform nuclei) and brainstem. |
Shy-Drager syndrome | CT: Progressive atrophy of the brainstem and cerebellum. MRI: Low signal on T2-weighted images in putamen nuclei equal to or more pronounced than in the globus pallidus. Atrophy of the brainstem and cerebellum. | Autonomic dysfunction in adults with orthostatic hypotension; cerebellar and extrapyramidal clinical signs. |
Neurodegeneration with brain iron accumulation | CT: Zones of low attenuation in the globus pallidus bilaterally. MRI: Low signal with or without areas of high signal on T2-weighted images in the globus pallidus bilaterally; no gadolinium enhancement. | Rare autosomal recessive metabolic disorder from mutations in the pantothenate kinase 2 (PANK2) gene with onset usually in childhood with progressive limb rigidity and gait dysfunction, dysarthria, and mental deterioration. Increased iron deposition and destruction of globus pallidus and substantia nigra bilaterally. |
Pelizaeus-Merzbacher disease | CT: Cerebral and cerebellar atrophy, slightly decreased attenuation in the cerebral white matter. MRI: Heterogeneous or diffuse high signal on T2-weighted images in cerebral white matter, with or without involvement of the cerebellum, brainstem; with or without low signal on T2-weighted images in basal ganglia, thalami; without gadolinium contrast enhancement; progressive cerebral and cerebellar atrophy. | X-linked (type 1) or autosomal recessive (type 2) leukodystrophy; five subtypes; deficiency of proteolipid component of myelin; presentation during neonatal period (type 2)/infancy (type 1) with abnormal eye movements, nystagmus, delayed psychomotor development; death in first decade; males > females. |
Lysosomal enzyme defects | Tay-Sachs disease: CT: Increased attenuation in the thalami and decreased attenuation in the white matter; progressive cerebral and cerebellar atrophy. MRI: Slightly increased signal on T2-weighted images in caudate nuclei, putamen, and thalami, with or without slightly increased signal on T2-weighted images in cerebral white matter. Slightly reduced diffusion in ventral thalamic nucleus, progressive cerebral and cerebellar atrophy. Neuronal ceroid lipofuscinosis: CT: Progressive cerebral and cerebellar atrophy. MRI: With or without low or high signal on T2-weighted images in caudate, putamen, and thalami; with or without high signal on T2-weighted images in white matter; typically without gadolinium contrast enhancement. Progressive cerebral and cerebellar atrophy. Mucopolysaccharidoses: CT: Zones of decreased attenuation in the cerebral white matter, with or without foci of low attenuation in the corpus callosum, cerebral white matter, and basal ganglia. MRI: Foci or diffuse zones of high signal on T2-weighted images in cerebral white matter; with or without foci of high signal on T2-weighted images in the corpus callosum and basal ganglia; perivascular spaces, cerebral cortical/subcortical infarcts, progressive cerebral atrophy, with or without macrocephaly, with or without communicating hydrocephalus, with or without meningeal thickening. | Functional defects (usually autosomal recessive) involving lysosomal catabolic enzymes. Tay-Sachs disease: functional hexosaminidase deficiency; neuronal ceroid-lipofuscinosis: lipofuscin deposits in cytosomes; mucopolysaccharidoses: autosomal recessive or X-linked disorders related to abnormal metabolism of mucopolysaccharides (Hurler, Hunter, Sanfilippo, and Morquio syndromes). Result in axonal loss and demyelination related to the accumulation of abnormal metabolites within cells. |
Disorders of amino acid metabolism | Phenylketonuria, propionic acidemia, methylmalonic aciduria, homocystinuria, ornithine transcarbamylase deficiency, leucinosis (“maple syrup” urine disease), and glutaric acidemia: CT: Zones with decreased attenuation in the cerebral and/or cerebellar white matter, with or without zones with decreased attenuation in the globus pallidi, putamina, caudate nuclei, thalami, and brainstem. MRI: High signal on T2-weighted images in cerebral and/or cerebellar white matter, with or without high signal on T2-weighted images in globus pallidi, putamina, caudate nuclei, thalami, and brainstem. | Autosomal recessive disorders involving defective enzymes regulating amino acid metabolism and mitochondrial function. These enzymatic defects can cause significant alteration of normal formation and maintenance of myelin. |
Basal ganglia calcifications | CT: Idiopathic calcifications in the basal ganglia can be seen in adults older than 30 y. Calcifications involving the basal ganglia in patients younger than or older than 30 y may be seen with hypoparathyroidism, pseudohypoparathyroidism, pseudo-pseudo-hypoparathyroidism, hyperparathyroidism, hypothyroidism, lead toxicity, Fahr disease, and neurodegeneration with brain iron accumulation. MRI: Low, intermediate, or high signal on T1- and T2-weighted images in basal ganglia; no abnormal contrast enhancement. | Signal of calcium deposition can vary depending on the size and configuration of deposits. Calcifications can also occur in basal ganglia secondary to endocrine abnormalities involving calcium (hypoparathyroidism), hypothyroidism, iron metabolism (neuro-degeneration with brain iron accumulation), prior inflammatory disease (toxoplasmosis, tuberculosis, cysticercosis, etc.), prior ischemic disease or prior toxic exposure (lead intoxication, carbon monoxide, etc.), and Fahr disease. |
Acquired hepatocerebral degeneration | CT: No zones with definite abnormal attenuation seen in the basal ganglia, with or without cerebral and cerebellar atrophy. MRI: High signal on T1-weighted images in basal ganglia; no abnormal contrast enhancement. | Signal abnormality related to hepatic dysfunction (alcoholic cirrhosis, hepatitis, portal-systemic shunts) possibly related to increased serum ammonia and manganese levels. The signal hyperintensity may reverse after liver transplantation. |
Huntington disease | Disproportionate atrophy of basal ganglia (caudate > putamen > cerebellum/brainstem). CT: Progressive atrophy of caudate and putamen bilaterally. MRI: Variable low signal (iron deposition) or high signal (gliosis) changes on T2-weighted images involving the putamen bilaterally; usually no abnormal contrast enhancement. | Adults: Autosomal dominant neurodegenerative disease related to abnormal segment (CAG repeats) of DNA on chromosome 4 involving the Huntington gene. Usually presents after age 40 y with progressive movement disorders (choreoathetosis, rigidity, hypokinesia); behavioral and progressive mental dysfunction/dementia. Juvenile Huntington disease also occurs in a small number of patients in the second decade. Patients present with rigidity, hypokinesia, seizures, and/or progressive mental dysfunction. |






















Lesions | CT Findings | Comments |
Congenital neuronal migration disorders | ||
Lissencephaly | Absent or incomplete formation of gyri and sulci with shallow sylvian fissures and “figure 8” appearance of brain on axial images, abnormally thick cortex, gray matter heterotopia with smooth gray-white matter interface. | Severe disorder of neuronal migration (weeks 7–16 of gestation) with absent or incomplete formation of gyri, sulci, and sylvian fissures. Associated with severe mental retardation and seizures, early death. Other associated CNS anomalies are dysgenesis of the corpus callosum, microcephaly, hypoplastic thalami, and cephaloceles. |
Pachygyria (nonlissencephalic cortical dysplasia) | Thick gyri with shallow sulci involving all or portions of the brain. Thickened cortex with relatively smooth gray-white interface may have areas of decreased attenuation in the white matter (gliosis). | Severe disorder of neuronal migration. Clinical findings related to degree of extent of this malformation. |
Gray matter heterotopia | Laminar heterotopia appears as a band or bands of gray matter within the cerebral white matter ( Fig. 1.7 , p. 8 ) . Nodular heterotopia appears as one or more nodules of gray matter along the ventricles (Fig 1.8, p. 8 ) or within the cerebral white matter (Fig. 1.9, p. 9 ) . | Disorder of neuronal migration (weeks 7–22 of gestation) in which a collection or layer of neurons is located between the ventricles and cerebral cortex. Can have a bandlike (laminar) or nodular appearance isodense to gray matter; may be unilateral or bilateral. Associated with seizures and schizencephaly. |
Schizencephaly (split brain) | Cleft in brain extending from the ventricle to cortical surface lined by heterotopic gray matter. The cleft may be narrow (closed lip) or wide (open lip). | Association with seizures, blindness, retardation, and other CNS anomalies (septo-optic dysplasia, etc.). Clinical manifestations related to severity of malformation. Ischemia or insult to portion of germinal matrix seen before hemisphere formation. |
Unilateral hemimegalencephaly | Nodular or multinodular region of gray matter heterotopia involving all or part of a cerebral hemisphere with associated enlargement of the ipsilateral lateral ventricle and hemisphere. | Neuronal migration disorder associated with hamartomatous overgrowth of the involved hemisphere. |
Tuberous sclerosis | Foci and/or confluent zones of decreased attenuation in cerebral white matter. | Nonmalignant lesions in white matter associated with tuberous sclerosis, consisting of areas of demyelination and/or dysplastic white matter along pathways of radial glial fibers during neuronal migration. |
Congenital dysmyelinating disorders | ||
Alexander disease | Zones with decreased attenuation involving the peripheral frontal white matter with progressive involvement of the white matter posteriorly and centrally (internal and external capsules); can show a marginal pattern of contrast enhancement; can be associated with increases in size and weight of brain tissue. | Sporadic leukoencephalopathy, also referred to as fibrinoid leukoencephalopathy, presents in first year of life with macrocephaly, progressive psychomotor retardation, resulting often in death during early childhood; also juvenile and adult forms. Caused by mutation of gene on chromosome 17q21 resulting in abnormal function and levels of the filament protein (glial fibrillary acidic protein [GFAP]). |
Canavan–van Bogaert–Bertrand disease | Zones with decreased attenuation involving the peripheral cerebral and cerebellar white matter diffusely with progressive involvement of the white matter centrally and subsequent atrophy, with or without involvement of the globus pallidus; typically no contrast enhancement; associated with increases in size and weight of brain tissue, enlarged N-acetylaspartate (NAA) peak on magnetic resonance hydrogen spectroscopy. | Autosomal recessive (usually occurs in Ashkenazi Jews), spongy degeneration disorder of the brain caused by deficiency of aspartoacylase (from abnormal locus on chromosome 17-short arm) resulting in N-acetylaspartic aciduria and deposits in brain and plasma; presents in infancy with macrocephaly, hypotonia, seizures, spasticity, and optic atrophy; death often occurs in second year. |
Pelizaeus–Merzbacher disease | Heterogeneous or diffuse decreased attenuation in cerebral white matter; initially involves the subcortical white matter with progression to the other remaining white matter; with or without involvement of cerebellum and brainstem; no contrast enhancement; progressive cerebral and cerebellar atrophy. | X-linked (type 1) or autosomal recessive (type 2) leukodystrophy; five subtypes; deficiency of proteolipid component of myelin; abnormality on chromosome Xq22; presentation during neonatal period (type 2)/infancy (type 1) with abnormal eye movements, nystagmus, and delayed psychomotor development; death in first decade; males > females. |
Metachromatic leukodystrophy | Symmetric diffuse zones of decreased attenuation in deep cerebral/periventricular white matter with progression of abnormal attenuation peripherally to involve the subcortical white matter; decreased attenuation involving the cerebellar white matter; no contrast enhancement; progressive atrophy. | Autosomal recessive disease involving the ARSA gene on chromosome 22q13.31-qter with deficiency of arylsulfatase A in lysosomes resulting in toxic accumulation of ceramide sulfatide (myelin breakdown product) in macrophages and Schwann cells. Three subtypes depending on onset: late infantile form (80%), juvenile form, and adult form. Progressive neurologic deterioration with peripheral neuropathy, gait disorders, and cognitive dysfunction leading to death. |
Childhood adrenoleukodystrophy | Zones with decreased attenuation usually in parieto-occipital periventricular white matter and corpus callosum; progression of abnormality to the remaining cerebral white matter, with or without contrast enhancement at regions of active demyelination/inflammation. | X-linked recessive leukodystrophy (males) involving chromosome Xq28 with functional deficiency of the peroxisomal enzyme acyl- coenzyme A (CoA) synthetase, resulting in abnormal metabolism and breakdown of very long chain fatty acids. These fatty acids accumulate in many tissues, including brain, with resultant demyelination, inflammation, gliosis, and necrosis. Onset 3 to 10 y, with psychomotor retardation, seizures, hypotonia, facial dysmorphism, progressive and deterioration. Also other subtypes: neonatal onset and adult onset. |
Krabbe disease | Symmetric confluent zones of decreased attenuation involving the periventricular white matter with progressive involvement toward the subcortical white matter; cerebral white matter involved > cerebellar white matter; no contrast enhancement; progressive cerebral atrophy. | Also known as globoid cell leukodystrophy, autosomal recessive disorder involving chromosome 14q24.3-32.1 with deficiency of lysosomal enzyme galactosylceramide β-galactosidase, resulting in destruction of oligodendrocytes/myelin production. Three subtypes: infantile (most common), late infantile, and adult onset. Seizures, psychomotor dysfunction, optic atrophy, and progressive neurologic deterioration leading to death. |
Vanishing white matter disease | Symmetric diffuse zones of decreased attenuation involving the cerebral white matter with progressive involvement toward the subcortical white matter; cerebral white matter involved > cerebellar white matter; no contrast enhancement; associated with increased brain size followed by progressive cerebral and cerebellar atrophy. | Also referred to as childhood ataxia with diffuse CNS hypomyelination; familial disease involving the translation initiation factor on chromosome 3q27. Progressive neurologic deterioration begins in childhood. |
Inherited metabolic disorders | ||
Lysosomal enzyme defects | Tay-Sachs and Sandhoff diseases: Increased slightly high attenuation in the thalami and decreased attenuation in the cerebral white matter followed by progressive cerebral and cerebellar atrophy. Neuronal ceroid-lipofuscinosis: Progressive cerebral and cerebellar atrophy; with or without zones of decreased attenuation in white matter; typically no contrast enhancement. Mucopolysaccharidoses: Poorly defined zones of decreased attenuation in white matter; cerebral and cerebellar atrophy, with or without foci of low attenuation in corpus callosum and basal ganglia (prominent perivascular spaces), cerebral cortical/subcortical infarcts, with or without macrocephaly, with or without communicating hydrocephalus, with or without meningeal thickening. | Functional defects (usually autosomal recessive) involving lysosomal catabolic enzymes. Tay-Sachs: functional N-acetyl hexosaminidase A deficiency; from mutation at HEXA gene on chromosome 15q-23-24. Sandhoffdisease: functional N-acetyl hexosaminidase A and B deficiency; from mutation at HEXB gene on chromosome 5q-13. Neuronal ceroid-lipofuscinosis is a relatively common progressive type of encephalopathy in children 125 000 births with lipofuscin deposits in cytosomes causing cerebral and cerebellar atrophy. Mucopolysaccharidoses: autosomal recessive or X-linked disorders related to abnormal metabolism of mucopolysaccharides (Hurler, Hunter, Sanfilippo, and Morquio syndromes) result in axonal loss and demyelination related to the accumulation of abnormal mucopolysaccharide (glycosaminoglycans) metabolites within cells of various organs. |
Disorders of amino acid metabolism | Phenylketonuria, propionic acidemia, methylmalonic aciduria, homocystinuria, ornithine transcarbamylase deficiency, citrullinemia, arginosuccinic aciduria, leucinosis (“maple syrup” urine disease, and glutaric aciduria): Zones with decreased attenuation in cerebral and/or cerebellar white matter, with or without zones with decreased attenuation in globus pallidi, putamen, caudate, thalami, and brainstem. | Autosomal recessive disorders involving defective enzymes regulating amino acid metabolism and mitochondrial function. These enzymatic defects can cause significant alteration of myelin formation. |
Mitochondrial metabolic disorders | MELAS and MERRF syndromes: Zones with decreased attenuation in the basal ganglia, usually symmetric, with or without decreased attenuation involving cerebral and cerebellar cortex; subcortical white matter abnormalities do not correspond to a specific large arterial vascular territory. | MELAS is a maternally inherited disease involving abnormal transfer of RNA-Leu from a mutation in mitochondrial DNA. MERRF is a mitochondrial encephalopathy associated with muscle weakness and myoclonic epilepsy, short stature, ophthalmoplegia, and cardiac disease. |
Leigh disease | Zones of low attenuation in both caudate nuclei and putamina, with or without decreased attenuation in white matter and cerebral cortex; typically no contrast enhancement. | Autosomal recessive disorder also referred to as subacute necrotizing encephalopathy; occurs in three forms: infantile, juvenile, and adult onset. Etiology related to abnormalities in oxidative metabolism in mitochondria from one of several defective enzymes (pyruvate dehydrogenase complex, cytochrome oxidase respiratory chain, adenosinetriphosphatase [ATPase], or complex I); progressive neurodegenerative disease. Lesions in brainstem are associated with loss of respiratory control. |
Kearns-Sayre syndrome | Zones of low attenuation in both caudate nuclei and putamina, with or without decreased attenuation in white matter, with or without calcifications in basal ganglia, thalami, and dentate nuclei; typically no contrast enhancement. | Mitochondrial disorder associated with external ophthalmoplegia, retinitis pigmentosa, and onset of clinical muscular and neurologic signs < 20 y. |
Cockayne syndrome | Zones with decreased attenuation involving the periventricular white matter, basal ganglia, and dentate nuclei, with calcifications in basal ganglia and dentate nuclei, progressive cerebral and cerebellar atrophy, microcephaly. | Autosomal recessive disorder with deficient repair mechanisms for DNA; presents in first decade with progressive neurologic dysfunction, cataracts, cutaneous photosensitivity, optic atrophy, and dwarfism. |
Toxic/metabolic | ||
Marchiafava-Bignami disease | Variable mixed low, intermediate, and/or high attenuation involving the corpus callosum, with or without other sites in cerebral white matter, with or without enhancement depending on stage of demyelination (acute, subacute vs chronic). | Acquired disorder associated with alcoholism and malnourishment, with demyelination, necrosis, or hemorrhage involving the corpus callosum, as well as other commissures and cerebral white matter. Symptoms include seizures, altered consciousness, ataxia, dysarthria, and hypertonia. |
Central pontine and extrapontine myelinolysis (osmotic myelinolysis) | Poorly defined zone of decreased attenuation involving the central portion of the pons (central pontine myelinolysis), Extrapontine myelinolysis occurs as zones with decreased attenuation in the cerebral white matter, external capsules, basal ganglia, thalami, midbrain, and middle cerebellar peduncles with or without occasional contrast enhancement. | Demyelinating disorder resulting from rapid correction of hyponatremia in chronically ill, malnourished, or alcoholic patients. Associated with diabetes mellitus, hepatitis, and chronic disease of the lungs, liver, and/or kidneys. |
Hypertensive encephalopathy (reversible posterior leukoencephalopathy) | Foci and/or confluent zones of decreased attenuation in subcortical white matter, with or without cerebral cortex, with or without contrast enhancement. Findings can be reversed if eliciting cause is corrected. | Occurs with elevations in blood pressure above the upper limit in cerebral vascular autoregulation resulting in capillary leakage of fluid in the brain often in arterial boundary zones. Associated with immunosuppressant drugs (e.g., tacrolimus/FK506 and cyclosporine), chemotherapy (e.g., cisplatin, and l-asparaginase), acute onset of hypertension, preeclampsia, eclampsia, renal dysfunction, and fluid overload. Neurologic symptoms include confusion, headaches, seizures, visual loss, dysarthria, and coma. Cortical abnormalities may be related to cortical laminar necrosis and hypoperfusion injury. |
Vascular | ||
Intracerebral hemorrhage | The attenuation of the hematoma depends on its age, size, location, hematocrit, hemoglobin oxidation state, clot retraction, and extent of edema. Hyperacute phase (4–6 h): Hemoglobin primarily as diamagnetic oxyhemoglobin (iron Fe2+ state). CT: High attenuation. Acute phase (12–48 h): Hemoglobin primarily as para-magnetic deoxyhemoglobin (iron Fe2+ state). CT: High attenuation in acute clot directly related to hematocrit, hemoglobin concentration, and high protein concentration. Hematocrit in acute clot approaches 90%. Early subacute phase (> 2 d): Hemoglobin becomes oxidized to iron Fe3+ state, methemoglobin, which is strongly paramagnetic. CT: High attenuation. Late subacute phase (> 7 d–6 wk): Intracerebral hematomas decrease 1.5 HU per day. Hematomas become isodense to hypodense; peripheral contrast enhancement from blood–brain barrier breakdown and vascularized capsule. Chronic phase: Hemoglobin as extracellular methemoglobin is progressively degraded to hemosiderin. CT: Chronic hematomas have low attenuation with localized encephalomalacia. Zones with high attenuation represent new sites of rebleeding. | Can result from trauma, ruptured aneurysms or vascular malformations, coagulopathy, hypertension, adverse drug reaction, amyloid angiopathy, hemorrhagic transformation of cerebral infarction, metastases, abscesses, viral infections (herpes simplex and CMV). |
Diffuse axonal injury | For acute injuries, one or multiple sites of hemorrhage are seen with high attenuation; commonly occur at the corpus callosum; cerebral cortical–white matter junctions, basal ganglia, and brainstem. | Brain injury caused by deceleration and rotational shear forces that results in disruption of axons and blood vessels. The greater the degree of axonal injury, the poorer the prognosis. |
Posthemorrhagic lesions | Zone or zones with decreased attenuation secondary to gliosis and encephalomalacia involving cerebral or cerebellar white matter, basal ganglia, thalami, and/or cerebral or cerebellar cortex; typically no contrast enhancement. | Sites of prior hemorrhage can have variable appearance depending on the relative ratios of gliosis, encephalomalacia, and blood breakdown products (methemoglobin, hemosiderin, etc.). |
AVM | Lesions with irregular margins that can be located in the brain parenchyma (pia, dura, or both locations). AVMs contain multiple tortuous vessels with or without calcifications. The venous portions often show contrast enhancement. Usually not associated with mass effect unless there is recent hemorrhage or venous occlusion. CTA can show the arterial, nidus, and venous portions of the AVMs. | Supratentorial AVMs occur more frequently (80%–90%) than infratentorial AVMs (10%–20%). Annual risk of hemorrhage. AVMs can be sporadic, congenital, or associated with a history of trauma. Multiple AVMs can be seen in syndromes Rendu-Osler-Weber (AVMs in brain and lungs and mucosal capillary telangiectasias) and Wyburn-Mason (AVMs in brain and retina with cutaneous nevi). |
Cavernous hemangioma | Single or multiple multilobulated intra-axial lesions with intermediate to slightly high attenuation, with or without calcification; typically show no contrast enhancement unless associated with a venous angioma. | Intra-axial vascular malformation composed of low-pressure endovascular-lined sinusoidal spaces often associated with sites of recent and/or prior hemorrhage; 80% are supratentorial. Can present with headache and/or seizures. Can be multiple in inherited syndromes. Supratentorial cavernous angiomas occur more frequently than infratentorial lesions. Can be located in many different locations; multiple lesions > 50%. Associated with venous angiomas and risk of hemorrhage. |
Venous angioma | No abnormality or small, slightly hyperdense zone on CT prior to contrast administration. Contrast enhancement seen in a slightly prominent vein draining a collection of small veins. | Considered an anomalous venous formation typically not associated with hemorrhage; usually an incidental finding except when associated with cavernous hemangioma. |
Ischemic disease related to occlusion of large vessels | CT features of cerebral and cerebellar infarcts depend on age of infarct relative to time of examination. Hyperacute phase (< 12 h): CT: No abnormality (50%); decreased attenuation and blurring of lentiform nuclei; hyperdense artery in up to 50% of cases. Acute phase (12–24 h): CT: Zones with decreased attenuation in basal ganglia, blurring of junction between cerebral cortex and white matter, sulcal effacement. Early subacute phase (24 h–3 d): CT: Localized swelling at sites with low attenuation involving gray and white matter (often wedge shaped), with or without hemorrhage. Late subacute phase (4 d–2 wk): CT: Localized swelling increases, then decreases; low attenuation at lesion can become more prominent; with or without gyral contrast enhancement. 2 weeks to 2 months: CT: With or without gyral contrast enhancement; localized mass effect resolves. > 2 months: CT: Zone of low attenuation associated with encephalomalacia. | Vascular occlusion of large arteries may be secondary to atheromatous arterial disease, cardiogenic emboli, neoplastic encasement, hypercoagulable states, dissection, or congenital anomalies. Cerebral infarcts usually result from arterial occlusion involving specific vascular territories, although occasionally they occur from metabolic disorders (mitochondrial encephalopathies, etc.) or intracranial venous occlusion (thrombophlebitis, hypercoagulable states, dehydration, etc.) that do not correspond to arterial distributions. |
Ischemic disease related to occlusion of small vessels | Multiple foci and/or confluent zones of decreased attenuation involving the subcortical and periventricular cerebral white matter, basal ganglia, and brainstem; no associated mass effect; typically no contrast enhancement. | Lesions in white matter and/or brainstem related to occlusive disease involving perforating arteries associated with hypertension, atherosclerosis, diabetes, vasculitis, and aging. Unlike multiple sclerosis, ischemic small vessel disease does not usually involve the corpus callosum because of its abundant blood supply from multiple branches arising from the adjacent pericallosal arteries. |
Periventricular leukomalacia | Multiple foci and/or confluent zones of decreased attenuation involving the subcortical and periventricular white matter, basal ganglia, and brainstem; no associated mass effect; no contrast enhancement; irregular ventricular margins and ventricular enlargement related to cerebral volume loss. | Ischemic injury involving fetal brain/premature infants with gliosis and resultant encephalomalacic changes involving periventricular white matter (fetal watershed vascular zones). Associated with neurologic deficits depending on severity of injuries and cerebral palsy. |
CADASIL | Multiple zones of decreased attenuation involving the subcortical and periventricular white matter, basal ganglia, thalami, and brainstem; no associated mass effect; no contrast enhancement. | CADASIL is an inherited abnormality involving chromo-some 19q12, which results in angiopathy of small and medium-sized arteries. Symptoms and signs begin in the fourth decade with headaches, transient ischemic attacks, strokes, and subcortical dementia. |
Susac syndrome | Multiple zones (usually < 10 mm) with decreased attenuation in the cerebral white matter and within the central portion of the corpus callosum. Zones of low attenuation may also occur in the basal ganglia (two thirds of patients). Leptomeningeal contrast enhancement may be seen in one third of patients. | Small vessel vasculitis of unknown etiology resulting in arteriolar occlusion and microinfarction involving cerebral white matter, retina, and cochlea. Patients often have headaches, cognitive changes, confusion, and memory impairment. Female/male ratio 3:1; age range 16 to 58 y. |
Radiation injury/necrosis | Focal lesion with or without mass effect or poorly defined zone of low to intermediate attenuation, with or without contrast enhancement involving tissue (gray matter and/or white matter) in field of treatment. | Usually occurs from 4 to 6 months to 10 y after radiation treatment; may be difficult to distinguish from neoplasm. PET and magnetic resonance spectroscopy might be helpful for evaluation. Radiation treatment in combination with intrathecal methotrexate may also result in necrotizing leukoencephalopathy. |
Inflammation | ||
Demyelinating Disease: multiple sclerosis, ADEM | Lesions located in cerebral or cerebellar white matter, corpus callosum, brainstem, and middle cerebellar peduncles. Lesions usually have low to intermediate attenuation with or without contrast enhancement. Contrast enhancement can be ringlike or nodular, usually in acute/early subacute phase of demyelination. Lesions rarely can have associated mass effect simulating neoplasms. | Multiple sclerosis is the most common acquired demyelinating disease usually affecting women (peak ages 20–40 y). Other demyelinating diseases are acute disseminated encephalomyelitis/immune mediated demyelination after viral infection; toxins (exogenous from environmental exposure or ingestion of alcohol, solvents, etc.); or endogenous from metabolic disorders (leukodystrophies, mitochondrial encephalopathies, etc.), radiation injury, trauma, or vascular disease. |
Sarcoidosis | Poorly marginated intra-axial zone with low to intermediate attenuation; usually shows contrast enhancement, with localized mass effect and peripheral edema. Often associated with contrast enhancement in the leptomeninges. | Multisystem noncaseating granulomatous disease of uncertain cause that can involve the CNS in 5% to 15%. Associated with severe neurologic deficits if untreated. |
Infection | ||
Cerebritis | Poorly defined zone or focal area of low to intermediate attenuation, minimal or no contrast enhancement; involves cerebral cortex and white matter in bacterial and fungal infections. | Focal infection/inflammation of brain tissue from bacteria or fungi, secondary to sinusitis, meningitis, surgery, hematogenous source (cardiac and other vascular shunts), and/or immunocompromised status. Can progress to abscess formation. |
Pyogenic brain abscess | Circumscribed lesion with low-attenuation central zone surrounded by a thin rim of intermediate attenuation representing the wall of the abscess, as well as a peripheral, poorly defined zone of low attenuation representing edema; ringlike contrast enhancement is seen at the wall of the abscess, which is sometimes thicker laterally than medially. | Formation of brain abscess occurs 2 weeks after cerebritis with liquefaction and necrosis centrally surrounded by a capsule and peripheral edema. Can be multiple. Complication from meningitis and/or sinusitis, septicemia, trauma, surgery, or cardiac shunt. |
Fungal brain abscess | Vary depending on organism; lesions occur in meninges and brain parenchyma; solid or cystic-appearing lesions with low to intermediate attenuation, nodular or ring pattern of contrast enhancement; peripheral low- attenuation edema in adjacent brain tissue. | Occurs in immunocompromised or diabetic patients with resultant granulomas in meninges and brain parenchyma. Cryptococcus involves the basal meninges and extends along perivascular spaces into the basal ganglia. Aspergillus and Mucor spread via direct extension through paranasal sinuses or hematogenously and invade blood vessels, resulting in hemorrhagic lesions and/or cerebral infarcts. Coccidioidomycosis usually involves the basal meninges. |
Encephalitis | Poorly defined zone or zones of low to intermediate attenuation, minimal or no contrast enhancement; involves cerebral cortex and/or white matter; minimal localized mass effect. Herpes simplex typically involves the temporal lobes/limbic system with or without hemorrhage; CMV usually in periventricular locations. HIV often involves the periatrial/periventricular white matter. | Infection/inflammation of brain tissue from viruses, often seen in immunocompromised patients (e.g., herpes simplex, CMV, HIV, and progressive multifocal leukoencephalopathy) or immunocompetent patients (e.g., St. Louis encephalitis, eastern or western equine encephalitis, and Epstein-Barr virus). |
Rasmussen encephalitis | Progressive atrophy of one cerebral hemisphere with poorly defined zones of decreased attenuation involving the white matter, basal ganglia, and cortex; usually no contrast enhancement. | Usually seen in children younger than 10 y; severe and progressive epilepsy and unilateral neurologic deficits: hemiplegia, psychomotor deterioration. Etiology: chronic slow viral infectious process possibly caused by CMV or Epstein-Barr virus. Treatment: hemispherectomy. |
Creutzfeldt-Jakob disease | Zones of decreased attenuation in putamen and caudate nuclei bilaterally, with or without zones of decreased attenuation in white matter and cortex. Typically no contrast enhancement; progressive cerebral atrophy. | Spongiform encephalopathy caused by slow infection from prion (proteinaceous infectious particle); usually seen in adults age 40 to 80 y; progressive dementia; < 10% survive more than 2 y. |
Reye syndrome | With or without diffuse edematous low attenuation involving basal ganglia, white matter, and cortex bilaterally. | Disorder of unknown etiology occurring in children usually younger than 16 y. Symptoms (vomiting, lethargy, seizures, and sometimes coma leading to death) occur during recovery from viral-like illness. Mortality ~20%. |
Lyme disease | Foci and/or confluent zones of decreased attenuation in cerebral and/or cerebellar white matter, with or without contrast enhancement. | CNS manifestations presumed to occur from immune-related demyelination from Lyme disease (infection by spirochete Borrelia burgdorferi) transmitted by ticks. |
Tuberculoma | Intra-axial lesions in cerebral hemispheres and basal ganglia (adults) and cerebellum (children): low to intermediate attenuation, central zone of low attenuation with a thin peripheral rim of intermediate attenuation; with solid or rim pattern of contrast enhancement; with or without calcification. Meningeal lesions: Nodular or cystic zones of basilar meningeal contrast enhancement. | Occurs in immunocompromised patients and in inhabitants of developing countries. Caseating intracranial granulomas via hematogenous dissemination; meninges > brain lesions. |
Toxoplasmosis | Single or multiple solid and/or cystic lesions located in basal ganglia and/or corticomedullary junctions in cerebral hemispheres, low to intermediate attenuation, nodular or rim pattern of contrast enhancement; with or without peripheral edema. | Most common opportunistic CNS infection in AIDS patients, caused by ingestion of food contaminated with parasites (Toxoplasma gondii). |
Parasitic brain lesions | ||
Cysticercosis | Single or multiple cystic lesions in brain or meninges. Acute/subacute phase: Low to intermediate attenuation, rim with or without nodular pattern of contrast enhancement; with or without peripheral edema. Chronic phase: Calcified granulomas. | Caused by ingestion of ova (Taenia solium) in contaminated food (undercooked pork); involves meninges > brain parenchyma > ventricles. |
Hydatid cyst | Echinococcus granulosus: Single or rarely multiple cystic lesions with low attenuation with a thin wall with low to intermediate attenuation; typically no contrast enhancement or peripheral edema unless superinfected; often located in vascular territory of the middle cerebral artery. Echinococcus multilocularis: Cystic (with or without multilocular) and/or solid lesions; central zone of intermediate attenuation surrounded by a slightly thickened rim of low to intermediate attenuation; with contrast enhancement. Peripheral zone of low attenuation (edema) and calcifications are common. | Caused by parasites E. granulosus (South America, Middle East, Australia, and New Zealand) and E. multilocularis (North America, Europe, Turkey, and China). CNS involvement in 2% of cases of hydatid infestation. |
Neoplastic | ||
Astrocytoma | Low-grade astrocytoma: Focal or diffuse mass lesion usually located in white matter with low to intermediate attenuation, with or without mild contrast enhancement. Minimal associated mass effect. Juvenile pilocytic astrocytoma subtype: Solid/cystic focal lesion with low to intermediate attenuation, usually with prominent contrast enhancement. Lesions located in cerebellum, hypothalamus, adjacent to third or fourth ventricle, brainstem. | Often occurs in children and adults (age 20–40 y). Tumors comprised of well-differentiated astrocytes. Association with neurofibromatosis type 1, 10-y survival; may become malignant. Common in children; usually favorable prognosis if totally resected. |
| Gliomatosis cerebri: Infiltrative lesion with poorly defined margins with mass effect located in the white matter, with low to intermediate attenuation; usually no contrast enhancement until late in disease. Anaplastic astrocytoma: Often irregularly marginated lesion located in white matter with low to intermediate attenuation, with or without contrast enhancement. | Diffusely infiltrating astrocytoma with relative preservation of underlying brain architecture. Imaging appearance may be more prognostic than histologic grade; ~2-y survival. Intermediate between low-grade astrocytoma and glioblastoma multiforme; ~2-y survival. |
Glioblastoma multiforme | Irregularly marginated mass lesion with necrosis or cyst, mixed low and intermediate attenuation, with or without hemorrhage, prominent heterogeneous contrast enhancement, peripheral edema; can cross corpus callosum. | Most common primary CNS tumor; highly malignant neoplasms with necrosis and vascular proliferation, usually seen in patients older than 50 y; extent of lesion underestimated by CT; survival < 1 y. |
Giant cell astrocytoma/tuberous sclerosis | Circumscribed lesion located near the foramen of Monro with mixed low to intermediate attenuation, with or without cysts and/or calcifications, with heterogeneous contrast enhancement. Poorly defined zones of decreased attenuation in the cerebral white matter, often in the periatrial regions. | Subependymal hamartoma near the foramen of Monro; occurs in 15% of patients with tuberous sclerosis < 20 y; slow-growing lesions that can progressively cause obstruction of CSF flow through the foramen of Monro; long-term survival usual if resected. |
Pleomorphic xanthoastrocytoma | Circumscribed supratentorial lesion involving the cerebral cortex and white matter; low to intermediate attenuation, with or without cyst (s), heterogeneous contrast enhancement, with or without enhancing mural nodule associated with cyst. | Rare type of astrocytoma occurring in young adults and children, associated with seizure history. |
Oligodendroglioma | Circumscribed lesion with mixed low to intermediate attenuation; sites of clumplike calcification, heterogeneous contrast enhancement; involves white matter and cerebral cortex; can cause chronic erosion of inner table of calvarium. | Uncommon slow-growing gliomas with usually mixed histologic patterns (astrocytomas, etc.). Usually seen in adults older than 35 y; 85% supratentorial. If low grade, 75% 5-y survival; higher grade lesions have a worse prognosis. |
Ganglioglioma, ganglioneuroma, gangliocytoma | Circumscribed tumor, usually supratentorial, often temporal or frontal lobes; low to intermediate attenuation; with or without cysts, with or without contrast enhancement. | Ganglioglioma (contains glial and neuronal elements); ganglioneuroma (contains only ganglion cells). Uncommon tumors, < 30 y, seizure presentation, slow-growing neoplasms. Gangliocytoma (contains only neuronal elements, dysplastic brain tissue). Favorable prognosis if completely resected. |
Ependymoma | Circumscribed lobulated supratentorial lesion, often extraventricular, with or without cysts and/or calcifications; low to intermediate attenuation; variable contrast enhancement. | Occurs more commonly in children than adults; one third supratentorial, two thirds infratentorial; 45% 5-y survival. |
Hamartoma/tuberous sclerosis | Cortical/subcortical lesions with variable attenuation: calcifications in 50% of older children; contrast enhancement uncommon. Subependymal hamartomas: Small nodules located along and projecting into the lateral ventricles; calcifications and contrast enhancement are common. | Cortical and subependymal hamartomas are nonmalignant lesions associated with tuberous sclerosis. |
Primitive neuroectodermal tumor | Circumscribed or invasive lesions; low to intermediate attenuation; variable contrast enhancement; frequent dissemination into the leptomeninges. | Highly malignant tumors located in the cerebrum, pineal gland, and cerebellum that frequently disseminate along CSF pathways. |
Dysembryoplastic neuroepithelial tumor | Circumscribed lesions involving the cerebral cortex and subcortical white matter; low to intermediate attenuation; with or without small cysts; usually no contrast enhancement. | Benign superficial lesions commonly located in the temporal or frontal lobes. |
Lymphoma | Primary CNS lymphoma: Focal or infiltrating lesion located in the basal ganglia, periventricular regions, and posterior fossa/brainstem; low to intermediate attenuation; with or without hemorrhage/necrosis in immuno-compromised patients; usually show contrast enhancement. Diffuse leptomeningeal contrast enhancement is another pattern of intracranial lymphoma. | Primary CNS lymphoma more common than secondary, usually in adults older than 40 y; B-cell lymphoma more common than T-cell lymphoma; increasing incidence related to number of immuno-compromised patients in population. CT features of primary and secondary lymphoma of brain overlap. Intracranial lymphoma can involve the leptomeninges in secondary lymphoma > primary lymphoma. |
Metastases | Circumscribed spheroid lesions in brain; can have various intra-axial locations, often at gray-white matter junctions; usually low to intermediate attenuation; with or without hemorrhage, calcifications, and cysts; variable contrast enhancement, often associated with low attenuation from axonal edema. | Represent ~33% of intracranial tumors, usually from extracranial primary neoplasm in adults older than 40 y. Primary tumor source: lung > breast > GI > GU > melanoma. |






























Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


