BONE ORIGIN
Bone Island (Enostosis) and Osteopoikilosis
Osteoid osteoma and Osteoblastoma
Ossifying Fibroma
Osteosarcoma
BONE MARROW ORIGIN
Plasmacytoma and Multiple Myeloma
Ewing’s Sarcoma
Lymphoma of Bone
Leukemia
CARTILAGE ORIGIN
Chondroma, Ollier’s Disease, and Maffucci’s Syndrome
Chondroblastoma
Chondromyxoid Fibroma
Osteochondroma and Hereditary Multiple Exostoses
Chondrosarcoma
FIBROUS, HISTIOCYTIC, AND FIBROHISTIOCYTIC ORIGIN
Fibrous Dysplasia
Nonossifying Fibroma and Fibrous Cortical Defect
Desmoplastic Fibroma
Fibrosarcoma and Malignant Fibrous Histiocytoma
SYNOVIAL ORIGIN
Pigmented Villonodular Tenosynovitis
Synoviochondrometaplasia
Synoviosarcoma
MUSCLE ORIGIN
Leiomyoma
Leiomyosarcoma
Rhabdomyosarcoma
FAT ORIGIN
Lipoma
Liposarcoma
VASCULAR ORIGIN
Hemangioma
Glomus Tumor
Lymphangioma
Angiosarcoma
NOTOCHORD ORIGIN
Chordoma
MISCELLANEOUS OR UNKNOWN ORIGIN
Aneurysmal Bone Cyst
Giant Cell Tumor
Simple Bone Cyst
Epidermoid Cyst
Interosseous Ganglion Cyst
Adamantinoma
Ameloblastoma
NONNEUROMUSCLOSKELETAL ORIGIN
Metastatic Bone Disease
Bone tumors are one of the most serious diagnostic possibilities in patients with musculoskeletal complaints. Bone tumors are categorized as either primary or secondary. Primary bone tumors arise from bone and related soft tissues directly in their site of involvement and may be either benign or malignant. Secondary bone tumors arise “secondary” to a primary lesion and also may be benign (e.g., secondary aneurysmal bone cyst arising in an area of past trauma) or malignant (e.g., bone metastasis from a lung carcinoma).
Benign tumors usually are designated as such by the suffix –oma (e.g., enchondroma) and generally are not regarded as cancers. Malignant primary tumors of bone and other connective tissues are designated by the use of the term, or suffix, sarcoma after the tissue type involved (e.g., osteosarcoma).
Metastatic bone disease describes a malignant tumor that secondarily seeds to bone, usually from a primary malignancy of the epithelial tissue (designated as carcinomas) of the lung, breast, prostate, kidney, liver, and so on. For instance, if the patient has a bronchogenic carcinoma that metastasizes to the thoracic spine, the lung lesion is the primary lesion and the thoracic spine lesion is bone metastasis of the lung lesion.
In general terms both benign tumors of bone and bone metastasis are many times more common than primary malignancies of bone. Primary malignancy of bone is uncommon. Malignancy in general is thought to affect about 1 in 3 people over their lifetime. Approximately 1.2 million Americans are diagnosed with a malignancy each year. By comparison, fewer than 1 in 100,000 people are diagnosed with a true primary malignancy of bone. In fact, the incidence of primary malignant bone lesions (excluding multiple myeloma) is estimated at 8 per million persons. 61 The most common sarcomas include osteosarcoma (35.1%), chondrosarcoma (25.8%), Ewing’s sarcoma (16.0%), chordoma (8.4%), and fibrosarcoma (5.7%). Because it arises from the plasma cell of bone marrow, many sources do not consider multiple myeloma as a primary malignancy of bone, instead naming osteosarcoma as the most common primary malignancy of bone. However, multiple myeloma is by far the most common primary malignancy of bone if considered in the group of primary bone malignancies. With an incidence of about 4.1 per 100,000135 it is considerably more common than osteosarcoma.
The evaluation of a bone tumor necessitates careful assessment of the patient’s history and application of clinical studies to develop a concise list of differential possibilities. Diagnostic imaging plays a major role in developing and narrowing this list. With advances in technology, biologic tissue assessments have added a valuable tool to the arsenal of the investigator. Therefore the diagnosis of tumors is accomplished along three dimensions of assessment: clinical (e.g., gender, age, symptoms), imaging (e.g., location, appearance), and pathologic (e.g., microscopic cell type and molecular assessment).
Imaging Modalities
Imaging studies should define the lesion, determine its location, grade its aggressiveness, decide if the lesion is limited to one bone (monostotic) or if multiple bones are involved (polyostotic), assess soft-tissue involvement, and identify the lesion’s matrix. Plain film radiography remains the chief imaging modality for the initial assessment of bone tumors. Sometimes the radiographic presentation reveals a lesion that is nonaggressive with classic characteristics, necessitating no further examination and leading to an immediate diagnosis. However, further assessment often is necessary when lesions are poorly defined or accompanied by significant clinical signs and symptoms.
Computed tomography (CT) provides direct thin axial slices of anatomy, allowing for a more detailed assessment of complex anatomy (e.g., spine) than can be accomplished with plain film. CT demonstrates calcification well and therefore is capable of demonstrating calcification within the lesion’s matrix and the cortical response of the lesion’s host bone. Plain film radiographs and CT scans of the chest help to assess the possibility of pulmonary metastasis, the presence of which may alter the treatment plan.
Although radionuclide bone scans lack specificity, they are sensitive to the presence of early disease and are widely used to assess the possibility of multiple lesions, a finding that substantially limits the diagnostic list of possibilities. There is one notable exception to the sensitivity of bone scans. Bone scans often are falsely negative in the presence of multiple myeloma and purely lytic lesions. In this and other cases it may be best to apply radiographic surveys or multiregional magnetic resonance imaging (MRI) studies.
MRI has the ability to demonstrate abnormality of the bone marrow and delineate extraosseous involvement. Replacement of normal marrow by pathologic processes (e.g., metastasis, multiple myeloma, osteomyelitis) is readily demonstrated and provides an early sign of disease. However, MRI remains inferior to both plain film and CT for detailing calcification, ossification, cortical destruction, and periosteal reaction. MRI is especially valuable to assess the neurologic impact of the lesion. Plain film radiographs efficiently provide information about the rate of growth and aggressiveness of a lesion but do not establish a histologic diagnosis with the same accuracy as a biopsy. On MRI studies, most bone tumors are dark on T1-weighted images and bright on T2-weighted images. Fibrous tissue, cortical bone, desmoids, and scar tissue are dark on both T1- and T2-weighted images. Hemangiomas, lipomas, and liposarcomas are bright on both T1- and T2-weighted images because of the blood components of these lesions.
The information offered by imaging is coupled with clinical data, laboratory studies, and possibly biopsy to identify the specific lesion present.
Laboratory Tests
Laboratory tests generally are less helpful than imaging studies to diagnose bone tumors. Benign bone tumors demonstrate normal laboratory values, and malignant tumors often demonstrate normal laboratory values. However, a few characteristic laboratory findings may be noted with malignancy. For example, increased serum calcium levels and increased hydroxyproline in the urine are associated with massive bone osteolysis, as seen in generalized lytic metastasis or multiple myeloma. Some osteosarcoma, osteoblastic metastasis, and other bone-proliferating malignancies often are accompanied by increased serum alkaline phosphatase levels. Multiple myeloma is associated with monoclonal immunoglobulins “M-spike” on serum electrophoresis, Bence Jones proteins in the urine, hyperglobulinemia (reversed albumin-globulin [A/G] serum ratio), elevated creatine, and decreased hematocrit levels.
Bone Biopsy
Bone biopsy is the removal of suspect tissue from the body for examination by a pathologist. In most circumstances a biopsy provides the most accurate diagnosis possible, typically more accurate than can be obtained by imaging. However, biopsy is not especially helpful to determine the lesion’s rate of growth and aggressiveness. The latter qualities are best assessed by conventional and specialized imaging modalities; therefore a biopsy and imaging studies are complementary, leading to an end diagnosis.
The accuracy of the biopsy results depends on which region of the lesion is collected, the skill of the person performing the biopsy, and the clinical circumstances in which the biopsy takes place. It is generally most accurate if the biopsy is taken from the most aggressive and viable portion of the lesions. This often necessitates an open biopsy approach. However, an open biopsy is associated with higher complication rates than is a closed (percutaneous) biopsy approach. 171 Open biopsy procedures may contaminate malignant cells in normal tissue, interfering with otherwise successful limb-sparing procedures. Closed biopsy procedures generally are safer, but may not produce enough tissue for accurate diagnosis. Biopsy of highly vascular lesions is cautioned generally, given the risk of massive hemorrhage.
Tumor Staging
Tumor staging is integral to patient management. Once a tumor is found, the extent of disease is defined along three parameters. The first parameter defines the size of the tumor (T) and whether it has invaded surrounding tissues. The second parameter examines the extent of lymph node involvement (N). Lastly, one must know whether the tumor has metastasized to other regions of the body (M). These three parameters define the “TNM” system of staging lesions. Although some variation in the stages exists depending on the source and type of tumor being staged, a general presentation of the TNM method is presented in Box 13-1.
BOX 13-1
Tumor Staging
Tumor staging is the process of characterizing the extent of the patient’s disease along three parameters: tumor (T), lymph nodes (N), and metastasis (M).
T: Indicates the size and regional involvement of the tumor
TX: Primary tumor cannot be assessed
T0: No evidence of a primary tumor
Tis: Carcinoma in situ (the tumor cells are restricted to the epithelial layer of the involved tissue)
T1: Localized tumor; diameter is 2 cm or less
T2: Localized tumor; diameter is 5 cm or less and mild involvement of same organ tissue
T3: Advanced tumor; diameter is greater than 5 cm and there is extensive involvement of same organ tissue
T4: Massive tumor; diameter is greater than 5 cm and there is involvement of nerves, blood vessels, bone, or other organs
N: Indicates the extent of lymph node involvement
NX: Regional lymph nodes cannot be assessed
N0: No evidence of metastasis to regional lymph nodes
N1: Small tumor in one lymph node
N2: Medium tumor in one or more lymph nodes
N3: Large tumor in one or more lymph nodes
M: Indicates the presence or absence of distant metastasis
MX: Distal metastasis cannot be assessed
M0: No evidence of distal metastasis
M1: Evidence of distal metastasis present
A tumor stage (I to IV) can be calculated once the T, N, and M parameters are determined. In general, the higher the tumor stage, the lower the patient’s chance of survival.
Stage I: T1 N0 M0
Stage II: T2 N1 M0
Stage III: T3 N0 M0, T1–3 N1 M0
Stage IV: T4 N0–1 M0, T0–4 N2–3 M0, T0–4 N0–4 M1
Signs and Symptoms
Clinically important lesions usually are detected secondary to the patient’s complaint of pain or attention to a palpable mass. Aggressive bone lesions generate signs and symptoms directly, secondary to bone and tissue destruction. Classically the bone pain experienced is more dramatic at night and unrelated to physical activity. The patient may exhibit fever, impaired mobility, and cachexia with aggressive lesions. In contrast, most benign lesions are clinically asymptomatic, but commonly are recognized after pathologic fracture. This concept has been termed traumatic determinism; that is, the presence of the benign lesion is only determined as a result of trauma and subsequent fracture. Less frequently asymptomatic bone lesions are detected serendipitously on radiographs obtained for unrelated reasons.
Tumor Discriminators
Many tumors and tumorlike conditions produce similar imaging findings. The following list of radiologic and clinical parameters assists in narrowing the usually broad number of pathologic possibilities for a given radiographic appearance. In addition, the following parameters assist in evaluating the aggressiveness and clinical importance of a lesion.
PATIENT AGE
The age of the patient is an important clue to assist the examiner in differentiating between lesions that may look the same but are unique to certain age groups. Conversely, the patient’s age may suggest a diagnostic possibility that would not otherwise be considered from the radiographic appearance because of an atypical presentation. Age is more helpful when the age range associated with the tumor is narrow. Given only the patient’s age, an examiner can determine which tumor is present with a high degree of accuracy. 69 (Tables 13-1 and 13-2 present the ages at which common benign and malignant tumors develop, respectively.) Generally with malignant tumors, Ewing’s sarcoma and osteosarcoma are present in children, and metastatic bone disease and multiple myeloma are found in patients over 40 years of age. Benign tumors are most common in patients 10 to 30 years of age, slightly younger for simple bone cysts and slightly older for lipomas and hemangiomas.
| Age (in years) | Tumor |
|---|---|
| 5 to 10 | Simple bone cyst |
| 10 to 20 | Chondroblastoma, nonossifying fibroma, osteoid osteoma |
| 10 to 30 | Aneurysmal bone cyst, chondromyxoid fibroma, osteoblastoma, osteochondroma |
| 15 to 35 | Enchondroma, osteoma |
| 20 to 40 | Giant cell tumor |
| 30 to 50 | Lipoma |
| 40 to 50 | Hemangioma |
| Age (in years) | Tumor |
|---|---|
| Less than 1 | Metastatic neuroblastoma |
| 1 to 30 | Ewing’s sarcoma, osteosarcoma |
| 20 to 40 | Giant cell tumor, parosteal osteosarcoma |
| 30 to 50 | Fibrosarcoma, malignant fibrous histiocytoma, primary lymphoma of bone |
| More than 40 | Chordoma, chondrosarcoma, metastatic bone disease, multiple myeloma |
LOCATION
Individual tumors often predispose to certain bones (Table 13-3). Even more suggestive is the longitudinal (diaphysis, metadiaphysis, metaphysis, and epiphysis) (Fig. 13-1) and transverse (central or eccentric medullary, cortical, periosteal, and parosteal) (Fig. 13-2) location within bone (Table 13-4 and Fig. 13-3).
| *This percentage reflects the portion of primary malignant tumors of bone, excluding multiple myeloma. | |||||
| Tumor | Frequency | Age (median) at diagnosis | Typical location | Radiologic features | Comments |
|---|---|---|---|---|---|
| Benign | |||||
| Aneurysmal bone cyst | Uncommon | 10–30 | Metaphysis of long bones; sometimes posterior arch of vertebrae | Widely expansile defect of the cortex with cortical buttressing at the region of expansion from the host bone and very thin cortices at outer edge of the lesion. The lesions are osteolytic and exhibit “a soap bubble” appearance | The lesion represents cavities filled with extravasated blood. Lesions may occur secondary to trauma, or concurrent to other tumors (e.g., giant cell tumor). Pain and swelling at the site of involvement |
| Bone island | Very common | Any age | Any location but skull | Homogenously radiodense focus with spiculated “brush” border | The lesion represents a hamartoma of bone. They are painless, usually small, and of no clinical significance except for their differentiation from blastic metastasis. Multiple bone islands are termed osteopoikilosis. Osteomas are similar lesions found in the skull |
| Chondroblastoma | Rare | 10–20 | Proximal femur, about the knee, and proximal humerus | Small (<5 cm) osteolytic subarticular lesion with a well-defined margin of sclerosis | A rare lesion composed of chondroblasts and immature cartilage. They appear in the secondary centers of enchondral ossification of skeletally immature patients |
| Chondromyxoid fibroma | Rare | 10–30 | Proximal tibia and fibula | Radiolucent, oval, eccentric lesion | Tumor involving the cartilage-forming connective tissue of marrow space |
| Enchondroma | Common | 15–35 | Small tubular bones of the hands and feet; however, possible in any enchondrally formed bone | Small round or oval cystic defects, typically with stippled matrix calcification and endosteal scalloping | Discrete islands of cartilage surrounded by layered bone. Enchondromas occur within a bone; periosteal chondromas occur next to the cortex and under the periosteum. Long bone lesions are more likely to be painful. Multiple enchondromas is termed Ollier’s disease, and if soft-tissue hemangiomas are also presenting, it is termed Maffucci’s syndrome. Solitary enchondromas have a malignant potential of <1%; this rate rises to 30% for Ollier’s disease and greater for Maffucci’s syndrome |
| Fibrous cortical defect | Very common | 4–8 | Well-marginated, eccentric cortex-based osteolytic defect smaller than 3 cm | Nonaggressive fibrous lesion of bone, usually healing spontaneously | |
| Fibrous dysplasia | Common | <20 | Femur, tibia, craniofacial bones, ribs, and pelvis | Wide variety in appearance depending on the bone involved. Long bones demonstrate a midly expansile medullary lesion that may be mildly sclerotic (“smoky” or “ground glass” appearance.). “Shepherd’s crook” deformity is a lateral bowing and coxa vara deformity occurring in the proximal femur. Individual lesions often exhibit a thick “rind” of marginal sclerosis | Nonneoplastic disturbance of bone maintenance resulting in fibrous tissue replacing normal bone. Albright’s syndrome is the triad of polyostotic fibrous dysplasia, precocious puberty, and cutaneous hyperpigmentations (café-au-lait spots) |
| Giant cell tumor | Common (18% of benign bone tumors) | 20–40 | Distal radius, distal femur, proximal tibia | Subarticular, osteolytic, mildly expansile, eccentric, no marginal sclerosis, and sometimes with internal septation | Tumor of the supportive tissues of bone marrow; composed of large mononuclear stromal cells and interspersed osteoclastic-like multinucleated giant cells. About 5% to 10% may be malignant, marked by pain, swelling, and aggressive radiographic appearance |
| Hemangioma | Common | 40–50 | Craniofacial bones and vertebral bodies | Osteolytic defect with characteristic coarsened, honeycomb trabeculae; or vertical striations (“corduroy cloth”) | Represent vascular lesion of bone; typically asymptomatic; rarely expanding vertebral lesion may relate to spinal stenosis |
| Nonossifying fibroma | Very common | 10–20 | Metaphysis of tibia and femur | Well-marginated, eccentric cortex-based osteolytic defect >3 cm. Malignant lesions show cortical breakthrough and soft-tissue mass | Nonaggressive fibrous lesion of bone, usually healing spontaneously |
| Osteoblastoma | Uncommon | 10–30 | Posterior elements of the vertebrae; less commonly in proximal femur and tibia | Geographic, osteolytic, eccentric, mildly expansile lesion | Same as osteoid osteoma; highly vascular connective tissue nidus with interlaced osteoid tissue. Lesions may be painful |
| Osteochondroma | Common | 10–30 | Metaphyses of long bones, mostly lower extremities. Hereditary multiple exostoses of the proximal femora and pelvis is common | Sessile or pedunculated (“coat hanger” exostosis or “cauliflower cap”) cartilage capped bony outgrowth that is continuous with underlying bone | Abnormal outgrowth of lateral portion of the growth plate; affects epiphyseal growth. Multiple osteochondromas are referred to as hereditary multiple exostoses (HME), and are associated with a higher rate of malignancy (up to 25%) compared with solitary lesions (1% to 2%) |
| Osteoid osteoma | 11% of benign bone tumors | 10–20 | Cortex of proximal femur or tibia; less commonly the posterior elements of the vertebrae | Small radiolucent nidus surrounded by radiodense reactive sclerosis | Highly vascular connective tissue nidus with interlaced osteoid tissue. Classically pain at night that is alleviated by aspirin. Spine lesions present on the concave side of a painful scoliosis. Low recurrence following excision |
| Simple bone cyst | Common | 5–10 | Proximal humerus and proximal femur | Oblong, central, expansile, radiolucent subepiphyseal (less frequently diaphyseal) osteolytic lesion. At times the partial internal septations may fracture and inferiorly migrate (“fallen fragment” sign) | Fluid-filled lesion of uncertain etiology; clinically silent unless fractured |
| Malignant | |||||
| Bone metastasis | Very common | >40 | Spine, ribs, skull, and pelvis; rarely distal to the knees and elbows | Individual lesions appear osteolytic, less commonly osteoblastic, and characteristically with no or small soft-tissue mass or periosteal reaction | The most common mechanism is hematogenous seeding from primary tumors; the breast gives rise to 70% of female lesions, and prostate 60% of male lesions. Advanced osteolytic destruction may exhibit increased serum calcium. Osteoblastic lesions may reveal increased levels of alkaline phosphatase |
| Multiple myeloma | Common | >40 (60) | Vertebrae, rib, innominate, femur | Osteopenia, “punch-out” lesions, “raindrop” skull | Malignant plasma cell proliferation; with abnormal laboratory findings of Bence Jones proteins, anemia, thrombocytopenia, Rouleaux formation, Mott cell |
| Osteosarcoma | 35.1* | Bimodal: first mode 1–30 (19); second mode in elderly | Metaphyses of long bones (distal femur, proximal tibia, and proximal humerus) | Osteolytic (25%), osteoblastic (50%), or mixed (25%) lesion with poorly defined margins, aggressive periosteal reaction (“onion skin” or Codman’s triangle), and soft-tissue mass | Aggressive lesion of mesenchymal bone-producing cells. Lesions are painful, rapidly expanding, with malignant potential usually requiring amputation |
| Chondrosarcoma | 25.8* | >40 (53) | Femur, innominate, humerus | Destructive lesions appearing as a central lesion in bone with stippled calcifications, and cortical scalloping, or as a peripheral lesion extending from the bone’s surface appearing as an exploded osteochondroma | Malignancy of mesenchymal cartilage-producing cells exhibiting varying degrees of malignancy. Lesions are painful and expanding with malignant potential. They may be primary malignant lesions of bone, or secondary to benign cartilage lesions (e.g., enchondroma, osteochondroma) |
| Ewing’s sarcoma | 16.0* | 1–30 (15) | Femur, innominate, vertebrae, humerus | Permeative osteolytic eccentric pattern of bone destruction, often presenting as a scalloped deformity of the diaphyseal cortex (“saucerization”); in flat bones it appears as a mildly expansile, soap-bubble, osteolytic defect | Small round cell malignancy of bone. The patient exhibits a tender, warm, swollen limb |
| Chordoma | 8.4* | 30–70 (59) | Skull, sacrum, and C2 vertebral body | Appear as midline lesions, characterized by the presence of bone destruction and a large soft-tissue mass; calcification is noted in up to 40% of lesions on plain films | Develops from remnant portions of the notochord; patients complain of pain and enlarging soft-tissue mass, and regional visceral and neurologic involvement, such as headaches and diplopia for sphenooccipital lesions, and bowel and bladder dysfunction for sacrococcygeal lesions. Radiation is applied to those lesions that cannot be resected |
| Fibrosarcoma and malignant fibrous histiocytoma | 5.7* | 30–50 (59) | Distal femur and proximal tibia | Lesions appear as osteolytic destruction in the metaphysis, often extending to the diaphysis marked by cortical destruction and soft-tissue mass | Malignancy of deep fibrous tissue; patient complaints of pain and tenderness in the region of involvement (usually around the knee) |
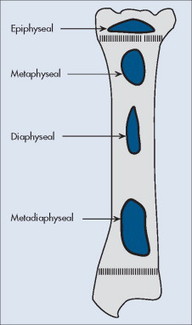 |
| FIG. 13-1 Longitudinal location within bone. |
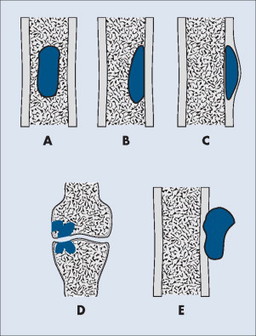 |
| FIG. 13-2 Axial location within bone. A, Central. B, Eccentric. C, Cortical. D, Intraarticular. E, Parosteal. (From Juhl JH, Crummy AB: Paul and Juhl’s essentials of radiologic imaging, ed 6, Philadelphia, 1993, JB Lippincott.) JB Lippincott |
| Location | Comments |
|---|---|
| Longitudinal | |
| Diaphysis | Round cell lesions (Ewing’s sarcoma, primary lymphoma of bone, multiple myeloma), malignant fibrous histiocytoma, adamantinoma |
| Diametaphysis | Osteoblastoma, chondromyxoid fibroma, nonossifying fibroma, Ewing’s sarcoma |
| Metaphysis | Aneurysmal bone cyst, chondrosarcoma, enchondroma, fibrosarcoma, giant cell tumor, osteosarcoma |
| Epiphysis | Chondroblastoma, giant cell tumor |
| Axial | |
| Central | Central chondrosarcoma, Ewing’s sarcoma, fibrous dysplasia, primary lymphoma of bone, solitary bone cyst |
| Eccentric | Aneurysmal bone cyst, fibrosarcoma, giant cell tumor, nonossifying fibroma |
| Cortical | Fibrous cortical defect |
| Parosteal | Parosteal chondrosarcoma, periosteal chondroma |
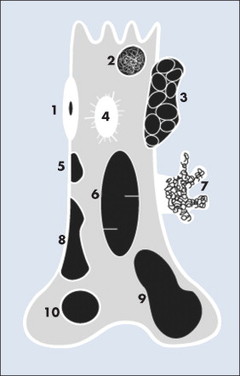 |
| FIG. 13-3 Location of common benign tumors of bone including: 1, osteoid osteoma; 2, enchondroma; 3, aneurysmal bone cyst; 4, bone island; 5, fibrous cortical defect; 6, simple bone cyst; 7, pedunculated osteochondroma; 8, nonossifying fibroma; 9, giant cell tumor; and 10, chondroblastoma. |
SOFT-TISSUE INVOLVEMENT
A soft-tissue mass, created when the tumor mass breaks through the host bone’s cortex, suggests an aggressive tumor. Soft-tissue masses related to tumors distort but do not disrupt intramuscular soft-tissue planes; soft-tissue masses secondary to infections may. Tumors tend to respect soft-tissue boundaries; infections may not.
HOST BONE REACTION
A number of pathologic processes are capable of accentuating or reviving normal mechanisms of bone growth resulting in periosteal or endosteal reactions. The appearance of the periosteal and endosteal reactions relates to the aggressiveness, intensity, and duration of the inciting process.
Periosteal and endosteal reactions must mineralize to be visible on radiographs; several patterns are identified (Fig. 13-4). Mineralization takes between 1 and 3 weeks. If the inciting process is indolent (e.g., vascular stasis), a thick, wavy periosteal reaction develops. Layered or lamellar periosteal reactions indicate a mildly aggressive underlying pathology (e.g., acute osteomyelitis). Aggressive pathology (e.g., osteosarcoma, Ewing’s tumor) may disrupt the periosteum, producing a radiating (sunburst) or parallel (hair-on-end) spiculated pattern (Fig. 13-5). The disrupted periosteum may form an acute angle with the cortex of the bone (Codman’s triangle) (see Fig. 13-4). Endosteal reactions are more limited, appearing thickened or scalloped in response to pathology within the medullary canal. Radiologic tumor grades based on the reaction of the host bone are presented in Table 13-5.
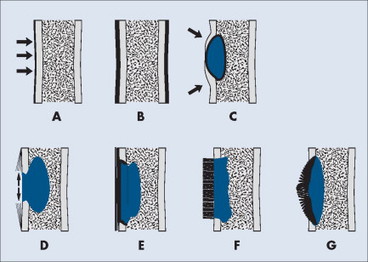 |
| FIG. 13-4 Periosteal reaction of bone. A, Thin lamellar. B, Thick lamellar. C, Cortical buttressing (arrows). D, Codman’s triangles (arrows). E, Aggressive lamellar. F, Hair-on-end spiculated. G, Sunburst. Patterns A through C are associated with benign lesions of bone. Patterns D through G are associated with aggressive lesions of bone (e.g., Ewing’s sarcoma and osteosarcoma). (From Juhl JH, Crummy AB: Paul and Juhl’s essentials of radiologic imaging, ed 6, Philadelphia, 1993, JB Lippincott.) JB Lippincott |
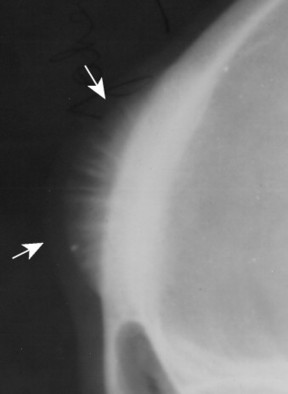 |
| FIG. 13-5 Skull lesion with a sunburst aggressive periosteal reaction (arrows). (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
| Aggressiveness | Characteristics |
|---|---|
| Low grade—nonaggressive | Geographic destruction surrounded by sclerotic rim of bone |
| Medium grade—moderately aggressive | Geographic destruction, short transition zone, possible sclerotic rim, possible bone expansion, possible thick periosteal reaction |
| High grade—highly aggressive | Permeative or moth-eaten destruction, wide transition zone, no surrounding sclerosis, possible bone expansion, aggressive periosteal reaction |
The tumor grade ultimately is derived from microscopic appearance of the tumor, as performed by a pathologist in association with the imaging findings and clinical data. Tumor staging (see Box 13-1) and grading relate inversely to the patient’s chance of survival.
PATTERN OF BONE DESTRUCTION
A number of explanations exist as to why tumors produce bone destruction. Tumors may stimulate osteoclastic activity by direct pressure, local hyperemia, or invasion. Bone destruction may be permeative, moth-eaten, or geographic (Fig. 13-6). Geographic lesions generally are less aggressive than the more subtle moth-eaten– pattern lesions, which generally are less aggressive than the nearly imperceptible permeative pattern.
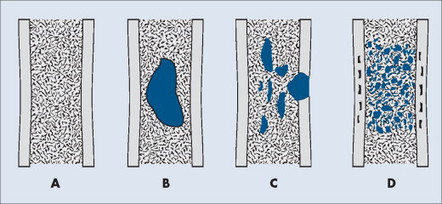 |
| FIG. 13-6 Patterns of bone destruction. A, Normal bone. B, Geographic. C, Moth-eaten. D, Permeative. (From Juhl JH, Crummy AB: Paul and Juhl’s essentials of radiologic imaging, ed 6, Philadelphia, 1993, JB Lippincott.) JB Lippincott |
An observer’s ability to recognize bone destruction depends primarily on the size and location of the lesion. Destructive lesions in cortical bone are more easily recognized than those in cancellous bone because greater contrast exists between osteolytic lesions and compact bone than osteolytic lesions and cancellous bone. Although technical factors should be considered, in general, lesions in cancellous bone that are less than 1 cm in diameter are difficult to recognize. Moreover, 30% to 50% of cancellous bone may be destroyed before an osteolytic lesion is recognized on the most optimally exposed and processed radiographs. 36,41,71,101 Osteolytic lesions in the diaphysis are more easily recognized because of the higher proportion of compact bone.
SIZE OF THE LESION
Each tumor has a unique growth rate influenced in part by the nature of the lesion and the response of the host bone. Markedly expansile lesions result when endosteal bone reabsorption of the inner cortex occurs in concert with periosteal appositional, intramembranous new bone growth of the outer cortex (Fig. 13-7). Although generally it is true that larger lesions are more aggressive than smaller ones, one should be careful not to judge a tumor’s clinical importance by size alone.
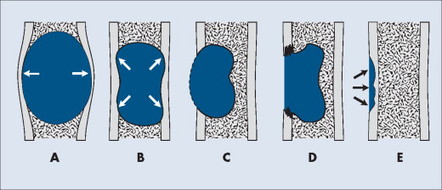 |
| FIG. 13-7 Cortical bone reaction. A, Expansile. B, Endosteal scalloping. C, Thin, nearly imperceptible. D, Destructive. E, Saucerization. (From Juhl JH, Crummy AB: Paul and Juhl’s essentials of radiologic imaging, ed 6, Philadelphia, 1993, JB Lippincott.) JB Lippincott |
RATE OF GROWTH
The rate of growth is a very important characteristic for assessing the aggressiveness of a lesion. Benign lesions grow slowly and have thick margins. Malignant lesions grow quickly and have less defined margins. Although highly important, the assessment of a lesion’s growth rate is difficult because of the usual lack of available serial studies.
MARGINATION AND ZONE OF TRANSITION
The appearance of the zone of transition between a lesion and the host bone is probably the single best indicator of a lesion’s aggressiveness. An abrupt transition from normal to abnormal is a feature of benignancy. A wide transition is a feature of malignancy. The zone of transition is a direct reflection of the lesion’s aggressiveness and the response of the host bone to the lesion infiltration (Fig. 13-8). Margination describes the presence and thickness of the rim around the lesion. Malignant tumors typically are nonmarginated, a feature of their wide zone of transition (Figs. 13-8 and 13-9). Conversely, a lesion is most likely benign if surrounded by a sclerotic rim of varying thicknesses producing a narrow zone of transition. The presence of a thick margin is always accompanied by a short zone of transition and represents an attempt of the host bone to surround and limit a lesion’s growth. However, a short zone of transition is not always accompanied by a thick margin (e.g., giant cell tumor). The zone of transition should not be used as the sole criteria for determining the aggressiveness of the lesion. Even a well-marginated lesion can prove to be malignant in selected clinical circumstances. For example, a painful lesion presenting with a narrow zone of transition in a 60-year-old patient who has a previous history of a primary tumor should be considered bone metastasis until proved otherwise.
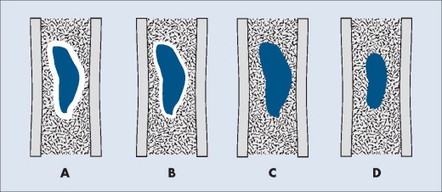 |
| FIG. 13-8 Margination of bone lesions. A, Thick. B, Thin. C, Absent. A through C demonstrate a short zone of transition between normal bone and the lesion. D, Ill-defined. Demonstrates a long zone of transition between normal bone and the lesion. A long zone of transition is most often associated with an aggressive lesion. (From Juhl JH, Crummy AB: Paul and Juhl’s essentials of radiologic imaging, ed 6, Philadelphia, 1993, JB Lippincott.) JB Lippincott |
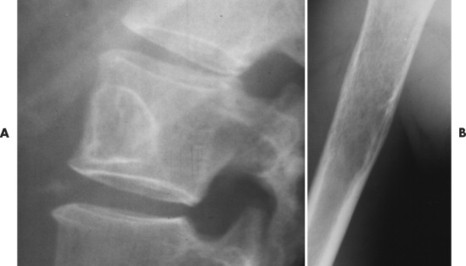 |
| FIG. 13-9 A, Well-defined benign defect of fibrous dysplasia characteristically exhibiting a thick surrounding margin and short zone of transition between the defect and the normal bone. B, Osteosarcoma with a wide zone between the center of the lesion and the normal bone. (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
TUMOR MATRIX
The matrix is the internal tissue, or substance, of a tumor. The radiographic appearance of the tumor’s matrix assists its categorization as primarily bone-, fibrous-, or cartilage-forming. Most bone tumors have a radiolucent matrix correlating to soft tissue. Only when the matrix is sufficiently mineralized with hydroxyapatite crystals will it become radiographically visible. Bone-producing tumors are radiodense. Highly aggressive bone-producing tumors appear less dense, with poorly formed osteoid material, than do nonaggressive bone-producing tumors. Tumors with a purely fibrous matrix appear radiolucent or slightly hazy (because of interspersed small bone fragments). Cartilage tumors usually are accompanied by irregular ringlike, flocculent, stippled, or flecklike radiodensities within the matrix (Figs. 13-10 and 13-11).
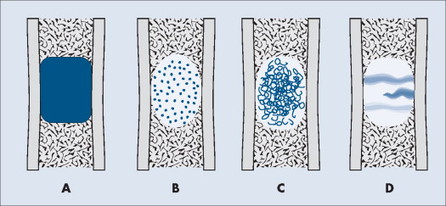 |
| FIG. 13-10 Matrix appearance of the bone lesion. A, Solid pattern of radiodensity indicates a bone matrix. B, Stippled appearance or, C, rings and arcs suggest a cartilage matrix. D, Hazy, smoky, or ground glass appearance correlates to a fibrous matrix of the lesion. |
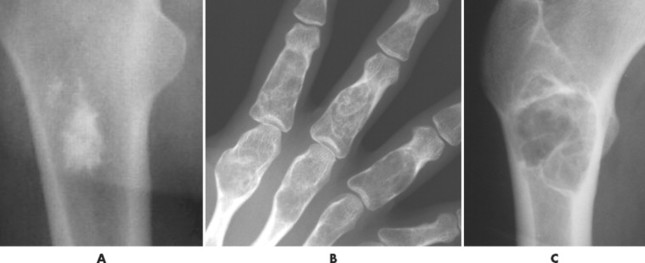 |
| FIG. 13-11 A, Radiodense appearance of bone matrix exhibited by a bone island. B, Arcs and curves of cartilage matrix exhibited by an enchondroma. C, Hazy or smoky appearance of fibrous matrix exhibited by a fibrous dysplasia. (B, Courtesy Gary Longmuir, Phoenix, AZ.) |
MULTIPLICITY
The presence of multiple lesions suggests different diagnostic possibilities than the presence of a single lesion. Generally radionuclide bone scanning determines if multiple lesions exist. Because radionuclide imaging is sensitive but not specific, plain film radiographs usually are taken at regions of increased radionuclide uptake. Radiographs usually are directed at evaluating a lesion rather than finding it. MRI and CT are applied less commonly to resolve uncertain findings of the radionuclide bone scan and plain films. The presence of multiple lesions limits the differential diagnosis. Common aggressive polyostotic diseases include metastatic bone disease and multiple myeloma. Less common possibilities include multicentric osteosarcoma and multifocal infections. Common nonaggressive polyostotic lesions include fibrous dysplasia, Paget’s disease, histocytosis, hereditary multiple exostoses (HME), multiple enchondromas (Ollier’s disease), and osteomyelitis.
Treatment Options
Treatment options include surgery, chemotherapy, radiation, or pain management; all may be applied alone or in combination. Surgical options extend from curettage for benign lesions to wide resection and possible amputation for highly malignant lesions. Each treatment approach is associated with complications, tumor recurrence, and varying success rates dependent on the patient’s clinical status and the type and stage of the lesions (Table 13-6). Often the treatment plan is aimed at offering the patient only palliative relief.
| Treatment | Description |
|---|---|
| Chemotherapy | Chemotherapy involves the intravenous or oral application of anticancer drugs Many agents are not selective to cancer cells, and may produce significant side effects (e.g., low blood count, vomiting, loss of appetite, loss of hair, mouth sores) |
| Complementary and alternative therapies | Complementary or alternative management strategies may be applied to reduce malignant lesions or offer palliative relief |
| Hormone therapy | The growth of some forms of cancer is enhanced by circulating hormones. For instance, estrogen promotes the growth of some breast cancers and testosterone promotes the growth of most prostate cancers. In an attempt to limit the growth of cancers, surgical removal of the ovaries or testes may be considered. More often drugs can be administered to limit the production of tumor-enhancing hormones or limit their effect on the tumor cells (e.g., tamoxifen) |
| Immunotherapy | Immunotherapy describes efforts to facilitate the patient’s immune system to recognize and destroy cancer cells by focusing on cytokines, monoclonal antibodies, and various tumor vaccines |
| Radiation therapy | Radiation therapy employs high-energy rays or particles to limit cancer growth, with the aim of either destroying the tumor, or at least shrinking its size, thereby giving the patient some palliative relief Radiation is most commonly applied via an external beam. Internal methods of delivering the radiation, for instance, the implantation of small seeds of radioactive material near the cancer, are less often used |
| Radiopharmaceuticals | Radiopharmaceuticals are radioactive substances most often applied to manage the bone pain of patients who have skeletal metastasis; agents include venous injections of strontium 89 (Metastron), samarium 153, and rhenium 186 |
| Surgical | Biopsy is performed, with either closed or open methods, to gather tissue specimens for laboratory evaluation. Curettage is the use of hand instruments to gently remove medullary bone. En bloc means the lesion is removed altogether as a whole. Wide resection implies that a surround margin of normal tissue is removed with the lesion. Limb-sparing surgery describes efforts to preserve limb function by the use of a metal endoprosthesis to reconstruct bone and joint function after wide resection. Amputation is the surgical removal of all or part of a limb or body part and is reserved for the most aggressive lesions |
Benign
Bone Island (Enostosis), Osteoma, and Osteopoikilosis
BACKGROUND
A bone island (enostosis) is a commonly encountered entity, representing a region of compact bone located in cancellous bone (Fig. 13-12). 128 Their etiology is not well documented, but most sources list them as developmental hamartomatous or dysplastic lesions. They may appear as a single defect or multiple defects with a variety of shapes. Bone islands are estimated to appear in 1% to 14% of the adult population without a demonstrated gender, age, or racial disposition.
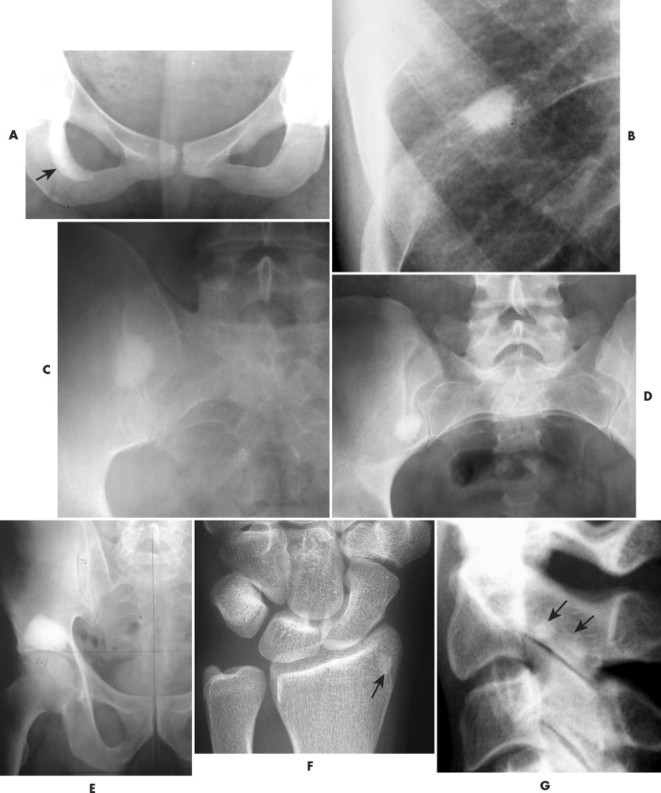 |
| FIG. 13-12 Well-defined focal radiopaque lesions representing bone islands of the A, pelvis (arrow); B, rib; C, D, and E, ilium; F, radial styloid (arrow); and G, spine (arrows). (E, Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
Osteopoikilosis (spotted bone disease) is a relatively uncommon condition of skeletal dysplasia that is marked by a symmetric presentation of multiple bone islands. Each lesion is microscopically identical to a bone island.
Osteomas differ from enostoses in that the former protrude from the surface of the affected bone and are found in the skull. Also, bone islands are encountered in all age groups, whereas osteomas typically are discovered in the adult patient. Multiple osteomas may be one component of Gardner syndrome, comprising colonic polyposis, osteomatosis, dental lesions, and soft-tissue tumors. 3,217
IMAGING FINDINGS
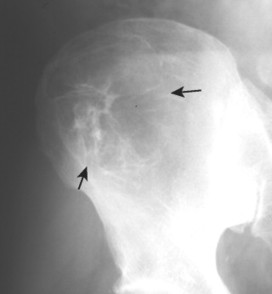
Enostoses are small (usually 0.1 to 2.0 cm), radiodense lesions that occur in all bones, although they are more common in the pelvis, proximal femora, and ribs. They have a characteristic thorny or spiculed border that blends into normal trabeculae, producing a typical whiskered or “brush border” periphery of the lesion (Fig. 13-13), a feature best seen on CT. Usually they are slightly oblong with their long axis running parallel to that of the host bone. Bone islands larger than 2 cm in diameter are less common, and are called giant bone islands. Larger lesions may exhibit active bone remodeling, evidenced by increased uptake of radiotracer with bone scintigraphy. 53 Although most bone islands are stable in size, some do exhibit slow growth over time. 19 Bone islands appear as hypointense signals on MRI.
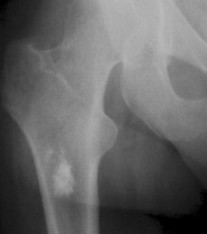 |
| FIG. 13-13 Bone island of the femur exhibiting a spiculated periphery that is known as a brush border. |
Osteopoikilosis exhibits a symmetric presentation common to the metaphyses of long bones, or commonly diffusely scattered in the carpals or tarsals. They may appear periarticular less commonly, especially about the hips (Fig. 13-14).
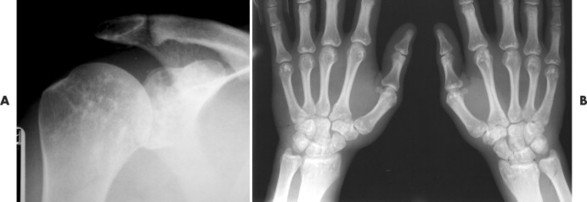 |
| FIG. 13-14 Multiple well-defined focal radiodense shadows distributed symmetrically near the joints representing multiple bone islands, known as osteopoikilosis of, A, the shoulder and, B, hands. (A, Courtesy Texas Chiropractic College, Pasadena, TX; B, Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
Osteomas appear as dense radiopacities found almost exclusively in the skull and facial bones, especially near the frontal sinuses (Figs. 13-15 and 13-16).
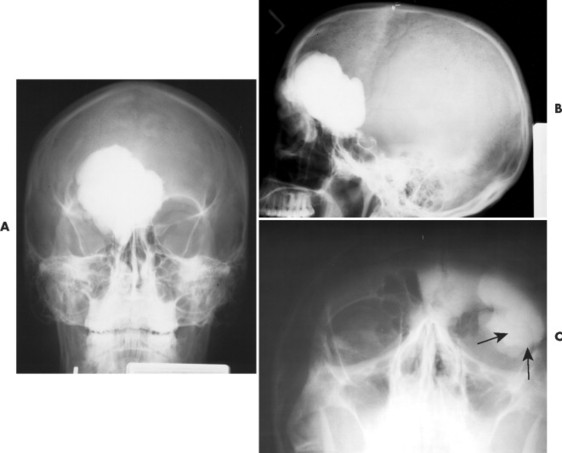 |
| FIG. 13-15 Large radiodense lesion of the skull consistent with an osteoma projected on, A, the anteroposterior; B, lateral; and, C, water’s (arrows) view. (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
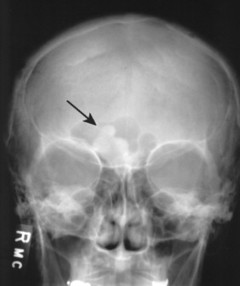 |
| FIG. 13-16 Irregular radiodense lesion of the right frontal sinus representing an osteoma (arrow). |
CLINICAL COMMENTS
A bone island usually is identified readily because it occurs without symptoms and has a characteristic appearance on imaging. However, if bone islands are multiple, large, growing, or present in patients with clinical red flags of aggressive bone disease (e.g., pain at night, personal history of past malignancy), differentiation from an entity such as osteoblastic metastasis may be warranted. Past radiographs may help in this differential. If past radiographs are unavailable, a bone scan represents the traditional method of differentiating bone islands from more aggressive entities. 96,183,215 However, it is possible that bone islands (especially large lesions) may appear active on bone scans, necessitating other methods of differentiation. MRI usually is definitive. Similar to a bone island, osteopoikilosis is not associated with clinical symptoms. Osteomas may become clinically significant if they enlarge and interfere with sinus drainage.
• Enostoses are common, usually small (<2 cm), and stable.
• Enostoses occur at all ages and in all bones; they are particularly common in the pelvis, femur, and ribs.
• Osteopoikilosis is a skeletal dysplasia of multiple bone islands.
• Osteomas are found in skull and facial bones, usually in adult patients.
Osteoid Osteoma and Osteoblastoma
BACKGROUND
An osteoid osteoma and osteoblastoma are histologically similar benign bone lesions that differ in size and (usually) radiographic appearance. Each consists of a highly vascularized nidus of connective tissue with interlacing trabeculae of osteoid and calcified bone surrounded by osteoblasts. Reactive bone response is variable and most often more pronounced on the cortical side of the lesions (less pronounced in osteoblastoma). Trabeculae merge into a mass of calcified bone toward the center of the lesion. Osteoid osteomas and osteoblastomas usually occur in patients between the ages of 10 and 25 years, with a 2:1 male predominance.
IMAGING FINDINGS
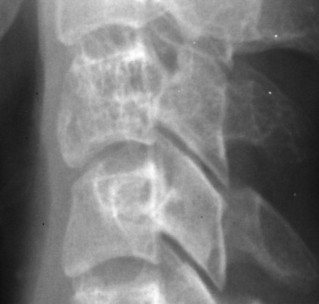
Osteoid osteoma appears as a small (<1.5 cm in diameter) osteolytic lesion with surrounding reactive sclerosis (Fig. 13-17). The central radiolucent nidus (FIG. 13-18FIG. 13-19 and FIG. 13-20) may be only a few millimeters in diameter and often contains a small focus of calcification. The surrounding reactive sclerosis may become so prominent that it completely obscures the radiolucent nidus, a feature that is particularly common when the lesions present in the vertebrae. The surrounding sclerosis represents a secondary finding and therefore may reverse after removal of the nidus. Osteoid osteomas are most commonly located in the cortex of long bones, usually the proximal femur (see Fig. 13-18) or tibia. Other locations include the posterior elements of the spine (e.g., concave side of painful scoliosis), humerus, hand, and talus. Osteoid osteomas usually are based in the cortex, but medullary, subperiosteal, and intracapsular locations do occur. 122
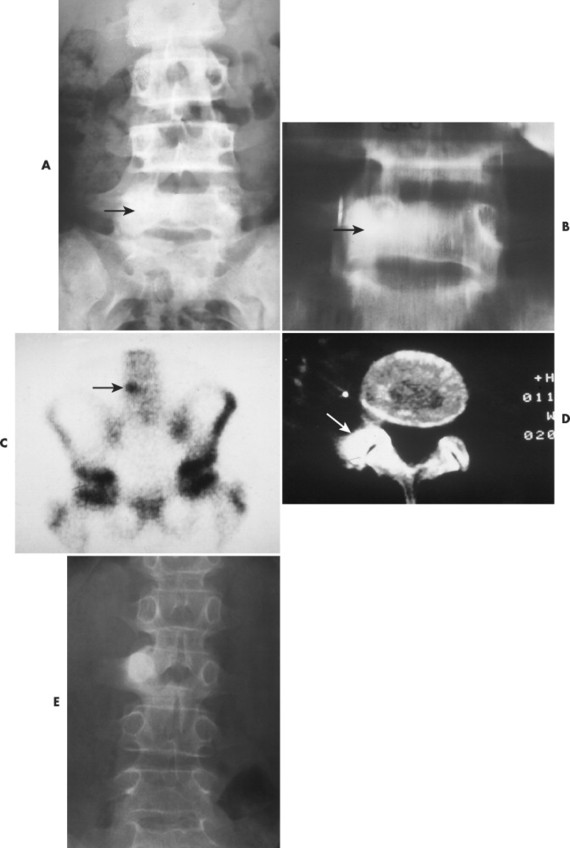 |
| FIG. 13-17 A, The pedicle is a predilectory site for the development of an osteoid osteoma of the spine (arrow). B, Tomogram manifesting the homogenous sclerosis associated with an osteoid osteoma, suggesting a “bright” pedicle (arrow). C, Increased radioisotopic uptake is noted in the presence of a pedicular osteoid osteoma (arrow). D, Computed axial tomographic scan exhibiting the presence of an osteoid osteoma in the vertebral arch (arrow). E, Note the dense, homogeneous sclerosis, representing the “bright” pedicle associated with an osteoid osteoma of the neural arch. (From Deltoff MN: The portable skeletal x-ray, St Louis, 1997, Mosby.) |
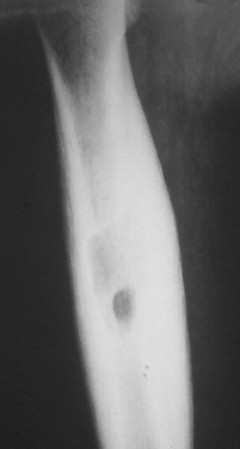 |
| FIG. 13-18 The femur represents another frequent site for the development of an osteoid osteoma. (From Deltoff MN: The portable skeletal x-ray, St Louis, 1997, Mosby.) |
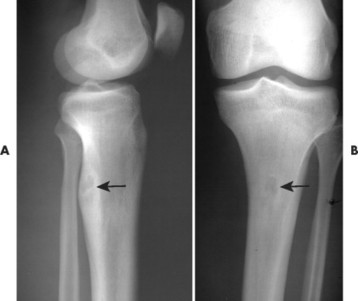 |
| FIG. 13-19 A and B, Osteoid osteoma presenting in the tibia (arrows). (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
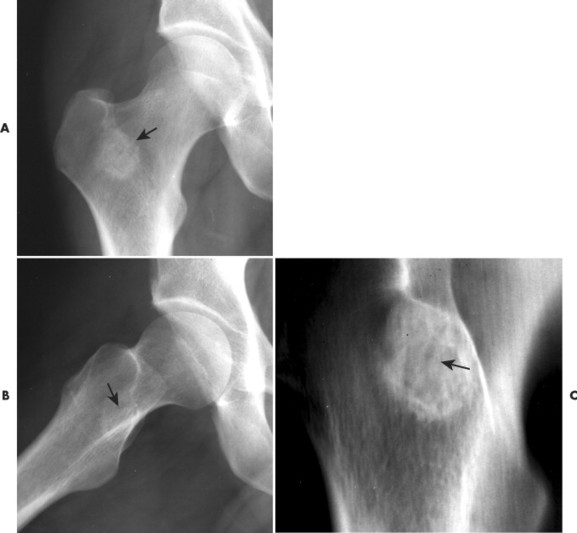 |
| FIG. 13-20 Osteoid osteoma of the intertrochanteric region of the proximal femur. The lesion appears as a radiodense region of reactive bone sclerosis surrounding a more radiolucent central nidus on, A, the anteroposterior; B, frog-leg; and C, linear tomogram (arrows). (Courtesy Gary Longmuir, Phoenix, AZ.) |
Osteoblastoma appears as a larger (≥1.5 cm in diameter), geographic, expansile, generally nonaggressive, eccentric medullary lesion. It may exhibit a sclerotic component. Osteoblastomas are most commonly located in the posterior elements of the spine157 (Fig. 13-21) and less frequently in the proximal femur, tibia, talus, ribs, and hands. 161
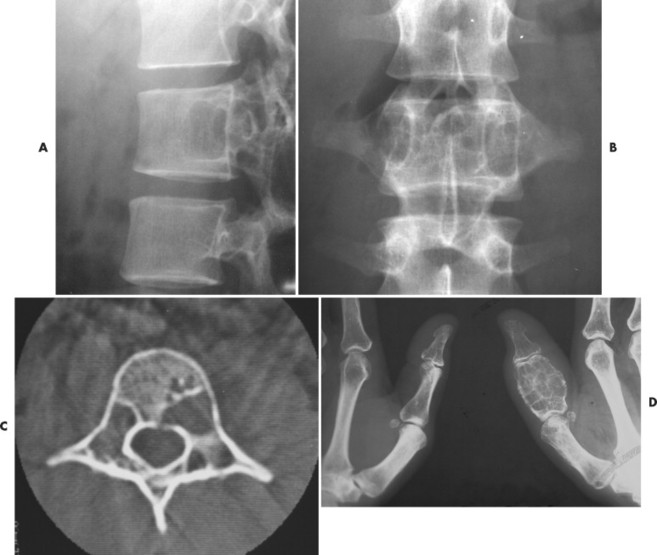 |
| FIG. 13-21 Expansile lesion of the neural arch and posterior body present on, A, the lateral; B, anteroposterior; and C, axial computed tomography image. The lesion is most consistent with an osteoblastoma. D, Another patient demonstrates similar bone expansion of the right thumb’s proximal phalanx, consistent with osteoblastoma. (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
Because osteoid osteomas and osteoblastomas are richly vascularized, an intense uptake is noted on radionuclide bone scans. CT is valuable to further delineate the lesions and demonstrate a radiolucent nidus. This is particularly true when imaging areas of complex anatomy, such as the spine.
On MRI the central nidus shows low to intermediate signal intensity on both T1- and T2-weighted MRI scans, and enhancement after contrast administration. 246
CLINICAL COMMENTS
Patients with osteoid osteomas and osteoblastomas often describe pain in the affected region. The pain begins as intermittent and vague, progressing to a dull and boring quality, which typically is not related to activity. Approximately 80% of patients with osteoid osteomas report a worsening of pain at night, which is characteristically alleviated by aspirin or other nonsteroidal antiinflammatory agents. 106,209
Osteoid osteomas of the spine typically are located on the concave side of a painful scoliosis, and should be excluded in all children and young adults who present with unexplained back pain or painful scoliosis. 124 Osteoblastomas also are associated with scoliosis but do not exhibit a predilection for the concave side. 134Markedly expansile osteoblastomas may cause canal stenosis. An osteoid osteoma in a cortical location of the medial femoral neck may mimic clinically an impeding stress fracture or infection. En bloc surgical excision of the nidus and some surrounding sclerosis is the preferred treatment for both lesions. Often the excision of the radiolucent nidus of an osteoid osteoma is followed by dramatic relief of the patient’s complaint. 170 Alternatively, long-term antiinflammatory treatment of 3 to nearly 4 years’ duration has been shown to regress the size of the lesion and provide permanent relief of symptoms. 129 Cryotherapy has been applied successfully as adjuvant treatment.
• Osteoid osteomas and osteoblastomas are histologically similar.
• Osteoid osteomas appear sclerotic, small (<1.5 cm), and cortically based in the femur or tibia, associated with pain that is dramatically alleviated with aspirin.
• Osteoid osteomas are most common to the lower extremities, particularly the neck of the femur.
• Osteoblastomas are larger (≥1.5 cm), expansile, medullary lesions of the spine, femur, and tibia.
• Both are noted in patients between 10 and 25 years of age.
Ossifying Fibroma
BACKGROUND
Ossifying fibromas are lesions that closely resemble fibrous dysplasia, both radiographically and pathologically. They present as two pathologically distinct lesions, one that occurs in the mandible derived from periodontal ligament (also termed cementifying fibroma) and a second that occurs in the tibia and fibula representing osteofibrous dysplasia. Osteofibrous dysplasia is marked by fibrous tissue and bone trabeculae circumscribed by osteoblasts, and occurring exclusively in the tibia and fibula. 31,125
Long bone ossifying fibromas typically occur within the first two decades of life and undergo a self-healing process until puberty and then stabilize. The mandibular lesions occur in the third and fourth decades and are much more common in females. 77 There is no gender predilection for the long bone presentation. The long bone lesions occur de novo or in association with an adamantinoma.
IMAGING FINDINGS
Whether they are in the mandible or long bones, ossifying fibromas initially present as radiolucent lesions, stabilize, and then become sclerotic over time (Fig. 13-22). In the long bones, lesions present in the tibia, fibula, or both. In fact, synchronous lesions of the tibia and fibula that appear on one side of the body are common presentations. Individual lesions appear radiolucent, circumscribed, expansile, and eccentric. Lesions in the tibia have a propensity to locate in the anterior or anterolateral cortex, creating an anterior bowing deformity of the bone’s shaft. The mandibular lesions likewise may deform the lower margin of the mandible and may contain a target, central opacification.
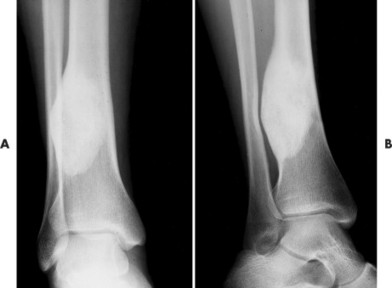 |
| FIG. 13-22 A and B, Ossifying fibroma of the distal tibia. (Courtesy Ian D. McLean, LeClaire, IA.) |
CLINICAL COMMENTS
Clinical concern for these rare lesions centers on differentiating them from typical fibrous dysplasia. Most lesions are treated conservatively.
• In long bones, ossifying fibromas are rare lesions of osteofibrous dysplasia occurring in the tibia or fibula during the first two decades of life.
• Ossifying fibromas initially appear radiolucent, becoming radiodense over time.
• Mandibular lesions typically are tissues in the molar or premolar area.
• Both lesions usually begin as painless masses and may exhibit bone deformity.
Malignant
Osteosarcoma
BACKGROUND
Osteosarcomas are highly aggressive malignant bone tumors. They are the second most common primary malignant bone tumors after multiple myelomas. However, because of their hematopoietic origin, some sources do not categorize multiple myeloma lesions as true bone tumors. In which case, osteosarcomas become the most common primary malignant bone tumors.
Although all osteosarcomas produce osteoid matrix, the proportion of chondroid and fibroid histologic constituents is variable. Only about 50% of osteosarcomas contain predominantly osteoid matrix, appearing osteoblastic on the radiographs. Osteosarcoma can be classified into intraosseous, surface, extraosseous, secondary, and multicentric types. 20
Intraosseous osteosarcoma.
Conventional and telangiectatic osteosarcomas are subtypes of the intraosseous classification. Conventional osteosarcomas account for 75% to 85% of all osteosarcomas. Conventional osteosarcomas arise within cancellous bone and then grow aggressively, extending into the medullary canal and penetrating the cortex to invade the soft tissues. Telangiectatic osteosarcoma is an aggressive, rare variant of osteosarcoma. It is characterized by large, blood-filled cavities and thin septations. 112
Surface osteosarcoma.
Periosteal and parosteal (juxtacortical) osteosarcomas are subtypes of surface osteosarcoma classification that arise from the cortex, subperiosteal tissue, or periosteum. They are marked by outward growth toward the soft tissues of the involved limb. Parosteal osteosarcoma is more common and exhibits large amounts of osteoid production. Periosteal osteosarcoma is rare and difficult to distinguish from periosteal chondrosarcoma because of the large amounts of chondroid and scant osteoid tissue that may be present in the lesion.
Other osteosarcomas.
Extraosseous osteosarcomas are soft-tissue tumors with the same histologic composition as conventional osteosarcomas. Secondary osteosarcoma most commonly occurs in elderly patients with Paget’s disease, infarcts, fibrous dysplasia, or regions of previous radiation therapy. 204,247 Multicentric osteosarcoma defines symmetrically distributed, multiple synchronous osteosarcomas of similar size and development. 190
Patient age.
Conventional and telangiectatic osteosarcomas have a slight male predominance and occur most commonly in people 10 to 25 years old; the median is 19 years. The occurrence of a second peak of incidence has been noted among those over the age of 60 years. 113 Periosteal osteosarcomas are seen most often in people 10 to 20 years old, and occur more commonly in female patients. Parosteal osteosarcomas occur in a slightly older age group than conventional or periosteal osteosarcomas. The average age of patients with extraosseous osteosarcomas is 45 years. Multicentric osteosarcoma occurs during the first decade.
IMAGING FINDINGS
Conventional osteosarcomas most commonly arise in the metaphysis of long bones, usually the distal femur (40%) (Fig. 13-23), proximal tibia (20%), humerus (9%), pelvis (5%), facial bones (5%), fibula (4%), and patella (<1%). Older patients exhibit a higher incidence of osteosarcoma in flat bones. The radiographic appearance of conventional osteosarcomas begins with a medullary osteolytic (25%), osteoblastic (25%), or mixed (50%) lesion in the metaphysis, often extending into the epiphysis or diaphysis. Ninety percent have a large, cloudlike density representing tumor bone and an aggressive (laminated, sunburst, “hair-on-end,” or Codman’s triangles) periosteal reaction (Fig. 13-24). The aggressive periosteal reaction occurs from tumor invasion lifting the periosteum of the bone, and subsequent reactive bone formation. A large soft-tissue mass is typical.
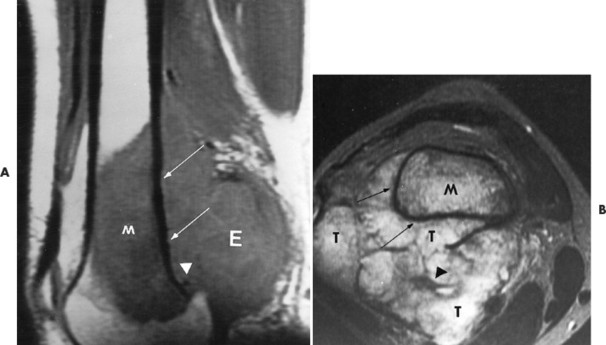 |
| FIG. 13-23 Distal femoral osteosarcoma. A, Sagittal T1-weighted magnetic resonance imaging demonstrates low signal intensity marrow involvement (M) with prominent posterior extension (E). B, Transaxial T2-weighted image reveals predominantly high signal intensity within both the marrow (M) and soft-tissue (T) components of the lesion. Areas of intact cortex (arrows) are evident on both images, along with tumor bone formation (arrowheads), suggesting the correct diagnosis. (From Sartoris DJ: Musculoskeletal imaging: the requisites, St Louis, 1993, Mosby.) |
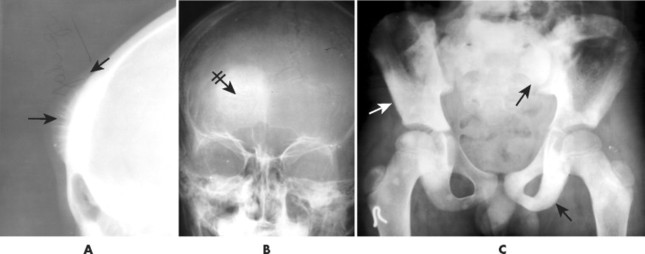 |
| FIG. 13-24 A and B, Osteosarcoma of the frontal bone with increased radiodensity (arrows) and aggressive periostitis (crossed arrow). C, Additional regions of increased radiodensity are noted in the pelvis (arrows), representing metastasis. (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
Telangiectatic osteosarcoma usually appears as an osteolytic, expansile lesion in the diaphysis of the femur, tibia, or humerus. It is characterized by periosteal reaction with a large soft-tissue mass and lack of visible bone production. The periosteal reaction often is layered, appearing as an “onion skin.” Parosteal osteosarcoma usually appears as a lobular, radiodense mass arising from a sessile base to surround the metaphysis of the host bone (FIG. 13-25FIG. 13-26FIG. 13-27FIG. 13-28 and FIG. 13-29). A thin radiolucent line is present but rarely seen, separating the tumor from the host bone. Nearly 70% of parosteal osteosarcomas are metaphyseal, arising from the posterior surface of the distal femur; other sites include the proximal tibia and humerus. Periosteal reaction is rare. Periosteal osteosarcomas present as soft-tissue masses extending from the diaphysis of long bones, usually the femur or tibia, which may have small perpendicular bone spicules or amorphous calcification within the tumor mass. The endosteal margin is not involved.
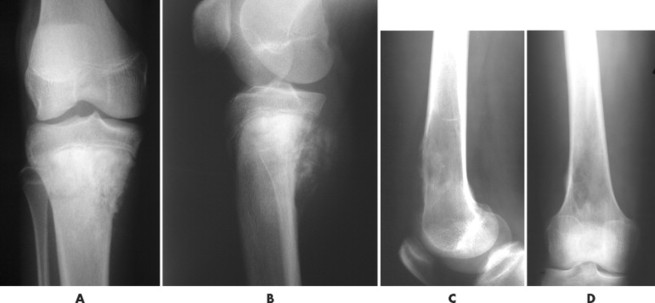 |
| FIG. 13-25 Osteosarcoma is most common around the knee. Two cases: A and B, the first presents osteosarcoma of the proximal tibia presenting the typical aggressive appearance and radiodense matrix. C and D, The second presents osteosarcoma of the distal femur appearing with a radiolucent destructive matrix. (C and D, Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
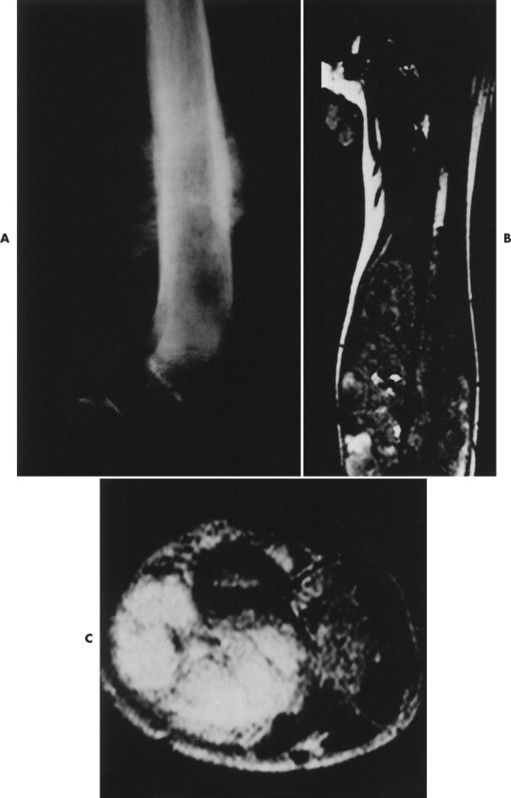 |
| FIG. 13-26 Characteristic appearance of osteosarcoma in the distal femur of a 15-year-old boy. A, Involved portion of bone has a mixed sclerotic and lytic appearance with periostosis and a soft-tissue mass. Note the sunburst appearance caused by radially oriented linear ossification. B, Coronal T1-weighted image demonstrates the relatively low signal tumor extending to the middiaphysis. The signal intensity of the tumor contrasts well with the normal bright signal of the yellow marrow. The rather low signal from the proximal diaphysis is caused by partial volume averaging of the cortical and medullary signal. The low signal in the proximal metaphysis is secondary to the presence of the normal red marrow. Note the loss of the signal void of the distal femoral cortex resulting from destruction (and replacement) by a tumor. The signal intensity of the tumor is only marginally different than the normal skeletal muscle. The bright signal areas represent areas of hemorrhage. C, T2-weighted image better defines the high signal intensity of the tumor from the surrounding low signal muscle vessels. (From Firooznia H et al: MRI and CT of the musculoskeletal system, St Louis, 1992, Mosby.) |
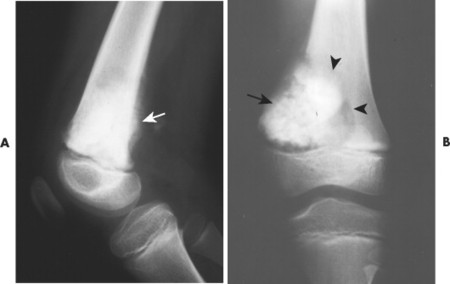 |
| FIG. 13-27 A, Osteosarcoma in the distal femur with aggressive spiculated periostitis (arrow) present in an 8-year-old boy. B, A band of osteolytic destruction (arrowheads) surrounding the radiodense matrix of the lesion (arrow) in the frontal projection. |
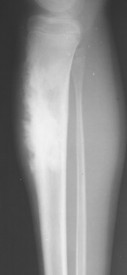 |
| FIG. 13-28 Aggressive periostitis outlining a radiodense matrix of a lesion within the proximal tibia in a 12-year-old patient. The lesion is consistent with osteosarcoma. (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
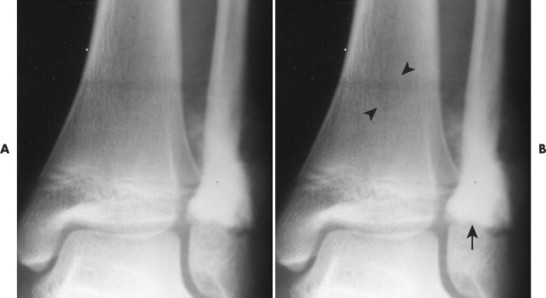 |
| FIG. 13-29 Osteosarcoma. A, Anteroposterior and, B, oblique ankle projections reveal an aggressive osteosclerotic lesion of the distal fibula. Aggressive periosteal and soft-tissue extension is noted on the medial side of the fibula (arrow). B, Incidentally noted is a nonossifying fibroma in the distal tibia, best seen on the oblique projection (arrowheads). |
Secondary osteosarcomas appear as destructive lesions usually indistinguishable from conventional osteosarcomas, occurring in the region of primary disease. Extraosseous osteosarcomas appear as large soft-tissue masses, most often in the buttocks and thigh. 79 More than half of the lesions demonstrate radiographic evidence of calcification. These lesions are best demonstrated by MRI. 234 Multicentric osteosarcoma is accompanied by multiple, symmetrically distributed radiodensities in the metaphyses.
For the most part, primary osseous lesions are well defined on conventional radiographs. CT and MRI are used to assess the extent of the lesion and evaluate its relationship to adjacent anatomy to aid surgical planning.
CLINICAL COMMENTS
Patients with osteosarcoma present with pain, swelling, and sometimes limited joint motion of the involved limb. Symptoms usually are present a few months before presentation. Serum alkaline phosphatase levels may be elevated. An initial complaint of pathologic fracture is more common with telangiectatic osteosarcoma. Multicentric, telangiectatic, and conventional osteosarcomas have poorer prognoses than surface osteosarcomas.
Conventional treatment includes the application of one or some combination of the following: excision, amputation, chemotherapy, and radiation. 52,164,194,227,244 Patient survival rates range from promising (with parosteal osteosarcoma) to poor (with multicentric osteosarcoma) and depend on the type of osteosarcoma, applied treatment, and general patient health status.
• Osteosarcoma is the second most common primary malignant tumor of bone after multiple myeloma.
• Osteosarcoma has a bimodal age distribution. Most lesions occur in patients between the ages of 10 and 25, with a later age group for the parosteal subtype.
• Osteosarcomas are aggressive, including bone, periosteal, and soft-tissue manifestations; 50% have radiodense matrices.
• These tumors most commonly arise from the metaphysis of long bones around the knee.
• Symptoms include pain, swelling, and limited motion of the limb that is involved.
Bone Marrow Origin
Malignant
Plasmacytoma and Multiple Myeloma
BACKGROUND
Multiple myeloma (MM) is a malignant monoclonal proliferation of plasma cells occurring predominantly in the red marrow of various bones. Plasma cells are mature B lymphocytes that produced immunoglobulins (antibodies). Therefore the proliferation of plasma cells results in increased amounts of circulating immunoglobulins (Ig) that have the potential to affect multiple organ systems.
Numerous diffuse foci or larger nodular accumulations of malignant plasma cells are surrounded by areas of increased osteoclastic activity. MM represents the most common primary tumor of bone, although some consider MM a hematologic disease, making osteosarcoma the most common primary tumor of bone. In any case, MM is the most common malignancy to present as a primary skeletal site. A solitary lesion of plasma cell myeloma is termed plasmacytoma (Fig. 13-30).
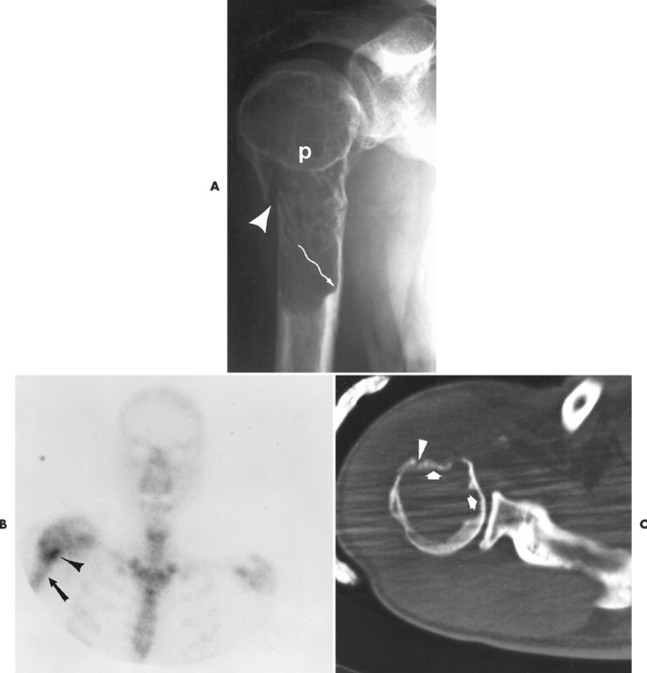 |
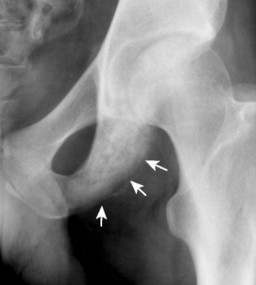
| FIG. 13-30 Plasmacytoma of the proximal humerus. A, Radiography reveals a septate lytic lesion (p) with endosteal scalloping (wavy arrow) and pathologic fracture (arrowhead). B, Increased activity is present within the lesion (arrow) and particularly at the site of fracture (arrowhead) as seen on a radionuclide bone scan. C, Computed tomography optimally demonstrates the degree of cortical thinning (arrows), as well as the fracture (arrowhead). (From Sartoris DJ: Musculoskeletal imaging: the requisites, St Louis, 1993, Mosby.) |
It is believed that the abnormal plasma cells produce osteoclastic-activating factors203 and osteoblastic inhibitory factors, which leads to increased bone resorption and decreased bone formation. 11,67 Initially MM invades the axial skeleton, progressing to the appendicular skeleton over time. It represents the most common primary malignant tumor of bone. Most patients with MM are 50 to 70 years old; rarely are those under the age of 30 years affected. 115 The median age is 60 years. A slight male predominance is often reported, and the lesions are twice as common among blacks as whites.
IMAGING FINDINGS
Approximately 80% of MM patients demonstrate radiographic features. Most commonly, MM occurs as multicentric (polyostotic) lesions of punched-out or moth-eaten patterns of bone destruction. Less commonly, MM presents as diffuse osteopenia without focal bone destruction; probably this is the most difficult presentation to recognize because it mimics osteoporosis. 34
Up to 30% of MM patients initially exhibit only a solitary lesion of bone known as a plasmacytoma (FIG. 13-131FIG. 13-132 and FIG. 13-133). Plasmacytomas appear as large (typically >4 cm in diameter), expansile, and cystic lesions. About 70% of patients with solitary plasmacytomas progress to multifocal disseminated MM. 243
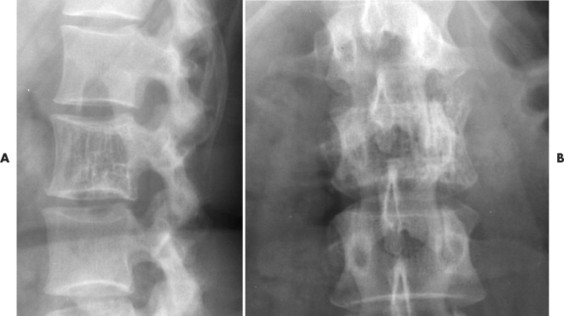 |
| FIG. 13-131 A, L3 segment has prominent vertical striations, a feature known as “corduroy cloth” or “jail bar” appearance of hemangioma. B, Anteroposterior projection has a coarsened appearance of the trabeculation. (Courtesy Gary Longmuir, Phoenix, AZ.) |
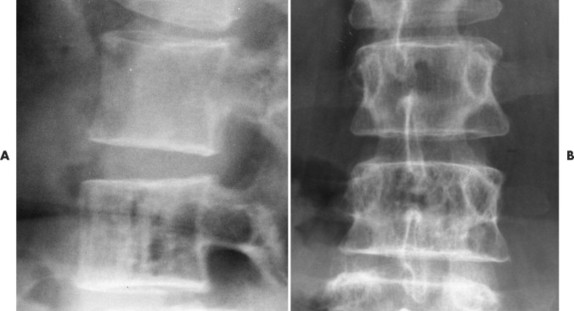 |
| FIG. 13-132 Hemangioma presenting with, A, prominent vertical striations and, B, coarsened trabeculation. |
 |
| FIG. 13-133 Hemangioma of C3 noted by coarsened trabeculation. (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
Rarely, MM presents as one or more sclerotic lesions, typically part of a larger syndrome known as POEMS (polyneuropathy, organomegaly, endocrinopathy, myeloma, and skin changes).
MM lesions exhibit a short zone of transition and no surrounding sclerosis, have no visible matrix (except for lesions in POEMS), and usually are less than 4 cm in diameter. MM originates in bones that are high in red marrow content. The skull, vertebral bodies, ribs, and proximal humerus and femur are most commonly involved (FIG. 13-34FIG. 13-35 and FIG. 13-36). Extraosseous manifestations are rare; they are found in fewer than 5% of patients. 95,177 Ribs and other small bones may be slightly expanded when involved.
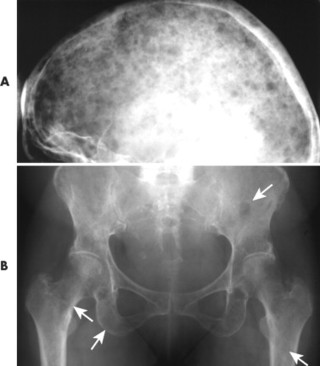 |
| FIG. 13-34 Multiple myeloma of, A, the skull and, B, pelvis. Both locations demonstrate the punched-out lesions characteristic of the disease (arrows). In the skull, the punched-out lesions give a raindrop skull appearance. |
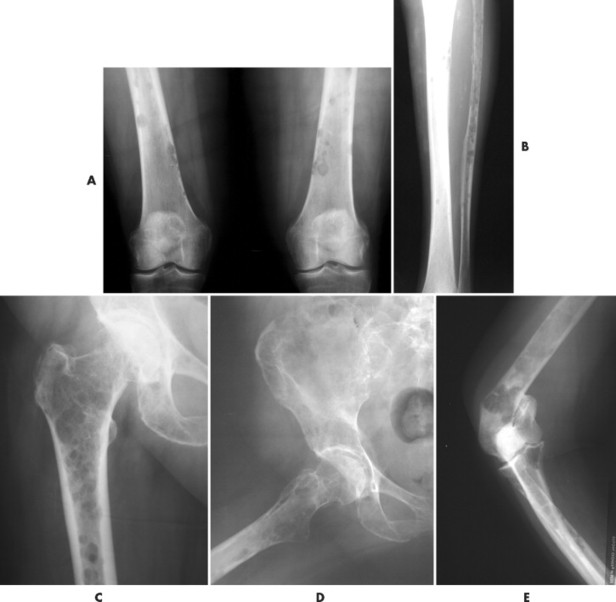 |
| FIG. 13-35 Multiple myeloma presenting with characteristic findings of osteopenia and “punched-out” lesions of, A, the femora; B, tibia and fibula; C, proximal femur; D, ilium; and E, elbow. (C, Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
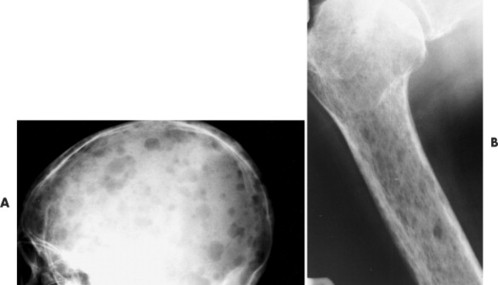 |
| FIG. 13-36 “Punched-out” lesions in, A, the skull (raindrop skull) and, B, proximal femur in different patients with multiple myeloma. (Courtesy Joseph W. Howe, Sylmar, CA.) |
Neoplastic plasma cells produce an osteoclastic activating factor that stimulates massive osteolysis with only minimal bone reaction; therefore MM frequently is associated with a negative bone scan. Plain film radiography appears more sensitive than radionuclide bone scanning; however, both miss a significant portion of cases. MRI is highly sensitive and is used in cases of negative radiographs and radionuclide imaging. 229
CLINICAL COMMENTS
Clinical findings are secondary to excessive plasma cell production. Pain is the most common patient complaint, reported in up to 70% of cases. Pathologic fractures are common and may be the presenting sign, especially collapsed vertebrae. MM is associated with multifocal bone pain (especially in the weight-bearing bones), amyloidosis, renal failure, hemorrhages, recurrent infections, and general weakness. 136 Amyloidosis and related organ impairment is seen in association with about 10% to 15% of MM patients. 130 Involvement of the kidneys and heart is most serious. In rare instances, patients with MM may remain asymptomatic. 59,60
Laboratory examination may reveal hypercalcemia, hyperuricemia, elevated creatine levels, anemia, and thrombocytopenia. The plasma cells can accumulate in renal cells, causing dysfunction and renal failure. The amount of Bence Jones proteins in the urine is an indicator of the degree of renal involvement. Elevated serum renal chemistries such as BUN (blood urea nitrogen) and creatinine point to renal impairment from the deposition of immunoglobulins in the kidney.
The corresponding serum abnormality consists of hyperglobulinemia with a reversal of the albumin-globulin (A/G) ratio. Most patients produce an overabundance of the immunoglobulin G (IgG) type. Serum abnormalities are recognized by electrophoresis and are considered central to the diagnosis of the disease. The increased levels of monoclonal immunoglobulins (M component) can be detected by protein electrophoresis as an abnormal “M spike” or “M component spike.” The level of M component reflects the tumor’s burden and can be used as a surrogate index to monitor the course of the disease and its therapeutic outcome. For example, 0.5 g/dl of M component in the serum estimates 10 g of neoplastic tissue in the body. High levels of M component are seen with MM and Waldenström macroglobulinemia. Usually MM patients who exhibit only an isolated plasmacytoma do not exhibit the M component abnormality in their serum.
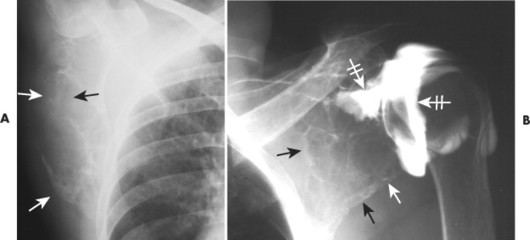
 |
| FIG. 13-32 A and B, Plasmacytoma presenting as an expansile cystic defect of the ilium in two patients (arrows). |
The peripheral blood smear reveals rouleaux formations, defined by red blood cells appearing stacked on top of one another like a roll of coins. This appearance is from the immunoglobulins, which act as “glue” and stick the red blood cells together.
Mott cells are found on bone marrow aspiration. Mott cells represent immature plasma cells with numerous intercellular vacuoles filled with immunoglobulins.
MM is diagnosed by a high index of clinical suspicion, with radiographic and laboratory correlation. A biopsy for histologic examination is necessary once a lesion is identified. Although variable, the usual course of the disease is that of gradual progression with a median survival of 3 years. 137 Treatment includes radiation and chemotherapy.
• Multiple myeloma (MM) is the most common primary malignant tumor of bone.
• Most patients are between 50 and 70 years of age.
• Bone scans are notoriously insensitive because of suppressed osteoblastic activity.
• MM exhibits a predilection for the axial skeleton.
• Most commonly, MM presents as multicentric (polyostotic) lesions of punched-out or moth-eaten patterns of bone destruction.
• Solitary lesions are termed plasmacytomas, appear less aggressive, and are expansile.
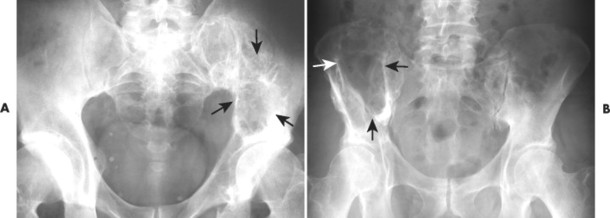
 |
| FIG. 13-33 Plasmacytoma of the ilium appearing as a cystic, radiolucent lesion with mild expansion (arrows). |
BACKGROUND
In 1921 James Ewing’s described a small, malignant round cell neoplasm of uncertain origin that bears his name. 78 Traditionally Ewing’s sarcoma was thought to originate from noncommitted mesenchymal cells; however, more recent studies suggest an association between Ewing’s sarcoma and a neuroectodermal origin. 55,150,248 Ewing’s sarcoma is the fourth most common primary malignant tumor of bone after multiple myeloma, osteosarcoma, and chondrosarcoma.
Ewing’s sarcoma of bone and soft tissue is one of four distinct histiologic subtypes of a group of lesions known as the Ewing’s family of tumors. The three other subtypes include peripheral neuroepithelioma (arising from the peripheral nervous system), esthesioneuroblastoma (developing from olfactive placode in the nasal vault), and Askin’s tumor (thoracopulmonary pediatric tumor).
IMAGING FINDINGS
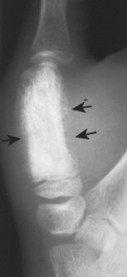
Ewing’s sarcoma may occur in any bone of the body, although the majority of cases occur in the pelvis and long bones of the lower extremities (FIG. 13-37FIG. 13-38FIG. 13-39 and FIG. 13-40). A diaphyseal location is classic, but a metadiaphysis is more common. 51,72
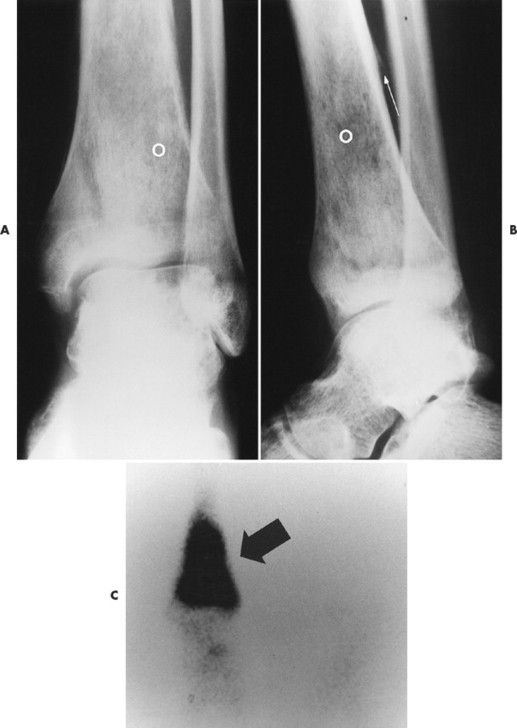 |
| FIG. 13-37 Ewing’s sarcoma. A and B, Frontal and lateral radiographs reveal permeative osteolysis (o) in the distal tibia with an associated Codman’s triangle (arrow). C, The process exhibits increased activity (arrow) on a radionuclide bone scan. (From Sartoris DJ: Musculoskeletal imaging: the requisites, St Louis, 1993, Mosby.) |
 |
| FIG. 13-38 Ewing’s sarcoma appearing as a mixed radiodense and osteolytic lesion of the ischial tuberosity with aggressive periostitis (arrows). (Courtesy Jack C. Avalos, Davenport, IA.) |
 |
| FIG. 13-39 Mixed radiodense and osteolytic lesion of the first metacarpal. Aggressive periostitis is noted (arrows) and is consistent with Ewing’s tumor. (Courtesy Ian D. McLean, LeClaire, IA.) |
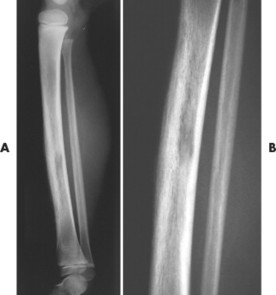 |
| FIG. 13-40 Ewing’s sarcoma appearing in the diaphysis of, A, the femur and, B, close-up. (Courtesy Ian D. McLean, LeClaire, IA.) |
The radiographic appearance is marked by ill-defined, permeative, osteolytic bone destruction, cortical erosion (known as saucerization), laminated or “hair-on-end” periosteal reaction, and a large soft-tissue mass. 199 Ewing’s sarcoma is less commonly a mixed osteolytic and sclerotic lesion (see Fig. 13-39). Only rarely does Ewing’s sarcoma appear as a predominantly sclerotic lesion. A Ewing’s tumor may appear similar to other round cell tumors, including lymphoma and metastatic neuroblastoma radiologically, but is differentiated from them easily with molecular probes and immunohistochemical techniques.
CLINICAL COMMENTS
Ewing’s sarcoma is very aggressive; usually patients experience a history of diffuse pain and swelling involving the affected region. Other symptoms frequently include an elevated sedimentation rate, erythema, leukocytosis, and fever, mimicking osteomyelitis, a main differential diagnosis. It has a high likelihood of metastasis, especially common to the pulmonary tissue. The prognosis has been very poor traditionally, but recent combinations of irradiation, surgical resection, and multidrug chemotherapy have elevated the 5-year survival to nearly 70%. 3 Survival rates are influenced by the age of onset, location, gender, and size of the lesion.
• Ewing’s sarcoma is the fourth most common primary malignant tumor.
• Patients are usually younger than 30 years of age.
• The clinical presentation may mimic an infection.
• Ewing’s sarcoma typically presents as a metadiaphyseal permeative osteolytic lesion with cortical saucerization and aggressive periosteal changes and less commonly appears as an osteosclerotic lesion.
Lymphoma of Bone
BACKGROUND
A lymphoma is a malignant tumor composed of lymphocytes, and (less commonly) histiocytes, that arise from lymph nodes, the spleen, or other sites of lymphoid tissue anywhere in the body. Lymphomas are classified by cell type, degree of differentiation, and nodular or diffuse pattern of distribution. Hodgkin’s disease (HD) is a distinctive form of lymphoma, separated from the larger, more common spectrum of non–Hodgkin lymphoma (NHL), by the presence of Reed-Sternberg cells. Reed-Sternberg cells are giant connective tissue cells, usually multinucleated, and characteristic of HD.
Lymphoma involves bone as either a primary focus or secondary to systemic lymphoma. Lymphoma is classified as a primary bone lesion if the single osseous lesion, with occasional minimal regional metastasis, is the only lymphoma lesion found in the body. Therefore primary lymphoma of bone is nonsystemic by definition. Primary lymphoma of bone is incorporated within the NHL classification and often is called reticulum cell sarcoma of bone. Primary NHL lymphoma of bone is microscopically identical to the nodal form of the disease; it differs only in its bone location. Burkitt’s lymphoma is a type of primary bone lymphoma. Secondary bone involvement can occur from either systemic NHL or HD. HD involves bone only as a secondary lesion.
IMAGING FINDINGS
Eighty-eight percent of patients with primary lymphoma of bone present with a single osseous lesion that fulfills the definition of primary NHL of bone (FIG. 13-41FIG. 13-42 and FIG. 13-43). 45 The remaining cases are polyostotic at presentation. Four percent of all patients with systemic NHL present with a concurrent skeletal lesion. Twenty percent of patients with HD demonstrate concurrent osseous lesions. 14,236
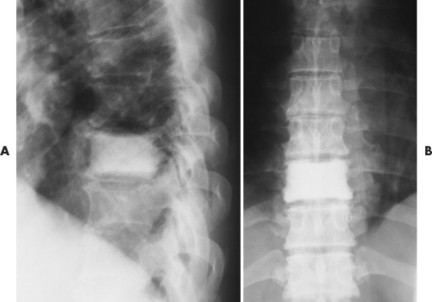 |
| FIG. 13-41 Lymphoma presenting as a radiodense “ivory vertebra” lesion of the lower thoracic spine in, A, the lateral and, B, anteroposterior projection. (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
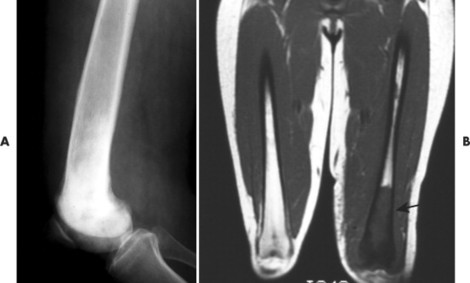 |
| FIG. 13-42 Lymphoma presenting as a sclerotic lesion in, A, the lower femur and demonstrating decreased marrow signal intensity on, B, the T1-weighted magnetic resonance imaging consistent with marrow replacement in the distal region of the left femur secondary to lymphoma (arrow). (Courtesy Ian D. McLean, LeClaire, IA.) |
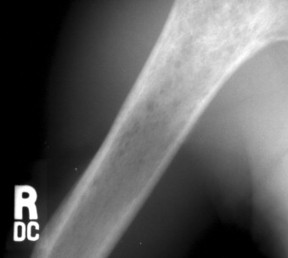 |
| FIG. 13-43 Lymphoma of bone appearing as a rarefied region of bone destruction in the proximal humerus. (Courtesy Joseph W. Howe, Sylmar, CA.) |
Osseous lesions caused by primary NHL of bone are radiographically indistinguishable from metastatic osseous lesions caused by systemic NHL and HD. 153 All types of lymphoma usually exhibit a permeative or moth-eaten pattern of osteolytic bone destruction (Figs. 13-42 and 13-43). In the long bones, the diaphysis is preferentially involved. Soft-tissue masses are common, and periosteal reactions are infrequent. 202 CT and MRI are useful to define the extent of the lesion, especially its soft-tissue component. Osteoblastic lesions are more common secondary to HD, often presenting as an “ivory vertebra” (Fig. 13-41).
Primary lymphoma of bone predominates in the lower extremities. Secondary (metastatic) bone involvement from systemic NHL and HD most commonly involves the spine, pelvis, and ribs. Burkitt’s lymphoma is most common in the mandible and maxilla (Fig. 13-44). 29
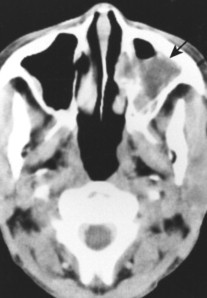 |
| FIG. 13-44 Computed tomography scan of a patient with Burkitt’s lymphoma. The scan demonstrates partial filling of the left maxillary sinus with thickening and irregularity of the medial sinus wall (arrow). (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
CLINICAL COMMENTS
Patients with osseous lymphoma typically complain of pain in the region of involvement but otherwise appear healthy. Radiation therapy, with or without adjuvant chemotherapy, may control the local bone lesions for years.
• Primary lymphoma of bone and secondary lymphoma of bone (systemic non–Hodgkin lymphoma and Hodgkin lymphoma) appear as permeative radiolucent lesions of the lower extremities, pelvis, and spine.
• Although generally osteolytic, bone lesions secondary to Hodgkin lymphoma are the most common to appear osteosclerotic (“ivory vertebra”).
• Lymphoma of bone occurs in patients over a wide age range.
BACKGROUND
Leukemia, the most common childhood cancer, has both acute and chronic forms. Although adults also may develop this cancer, the vast majority of leukemia patients are younger than 5 years of age. Lymphoblastic leukemia is the histologic variety in approximately 80% of cases in children.
IMAGING FINDINGS
Plain film findings are abnormal in nearly 50% of children with leukemia. The most common radiologic manifestation of this cancer is generalized osteopenia. The earliest radiographic findings are transmetaphyseal lucent bands near the physes of long bones such as the femur and tibia (Fig. 13-45). Rather than leukemic infiltrates, these bands appear to represent a metabolic reaction to the process. Alternatively, they may develop because pressure on the physis from the neoplastic infiltrates alters bony maturation at the zone of provisional calcification.
 |
| FIG. 13-45 Leukopenic lymphatic leukemia in a 2-year-old boy. A, Hand and forearm. B, Lower limbs; irregular destruction of all the bones is evident. Deep transverse zones of diminished density occupy the ends of the shafts. In addition to these destructive features, numerous large and small irregular patches of sclerosis are present, indicating a massive osteoblastic reaction. (From Caffey J: Pediatric x-ray diagnosis: an integrated imaging approach, St Louis, 1993, Mosby.) |
Discrete bony lesions may be present in the metaphysis or diaphysis, where they create a moth-eaten or permeative pattern of destruction. Periostitis also may develop with discrete bone lesions. In some cases, focal, spotty sclerosis is noted after chemotherapy or radiation treatment. Diffuse sclerosis of the bones has been encountered, but it is thought to be secondary myelofibrosis resulting from therapeutic interventions. 3
Infiltration of the meninges of the brain creates a separation of the skull sutures in a small percentage of patients with leukemia. Based on the radiographic findings alone, differentiation of infiltration of the meninges from neuroblastoma metastasis to the skeleton can be challenging because both conditions cause periostitis and separations of the sutures.
Focal accumulations of leukemic cells have been identified in a small number of patients with acute myelogenous leukemia. Because of their green color, these cells have been termed chloromas, and are more common in adults. Most commonly chloromas accumulate outside the skeleton. Skeletal accumulations most often develop in the face, sternum, and ribs. 16
CLINICAL COMMENTS
Patients with leukemia generally have a number of systemic complaints, including fever, malaise, and weakness. The musculoskeletal findings in leukemia include diffuse bone and joint pain, similar to the findings in juvenile rheumatoid arthritis. Joint and soft tissue swelling should alert the physician to the presence of a more serious condition. In leukemic patients the complete blood count (CBC) may be increased, normal, or decreased. However, the laboratory finding of blast cells in the peripheral blood smear is strongly suggestive of leukemia, and bone marrow biopsy is diagnostic.
• Leukemia is the most common childhood cancer.
• Radiographic findings include generalized osteopenia, transmetaphyseal lucent bands, and, occasionally, osteolytic bone lesions with a moth-eaten or permeative pattern of destruction.
• Clinical findings include fever, malaise, weakness, bruising, and joint pain.
Cartilage Origin
Benign
Chondroma, Ollier’s Disease, and Maffucci’s Syndrome
BACKGROUND
A chondroma is a cartilage tumor involving enchondrally formed bones. If the chondroma is located within the medullary canal, it is called an enchondroma (Fig. 13-46). Less commonly it is found adjacent to the cortex beneath the periosteal membrane, known as a periosteal chondroma, juxtacortical chondroma, or perichondroma. Chondromas represent discrete islands of hyaline cartilage surrounded by lamellar enchondral bone. Enchondroma is the most common tumor occurring in the phalanges and is the second most common benign cartilage neoplasm of bone after osteochondroma. 91 Enchondromas most often present in patients 20 to 30 years old, with an equal gender distribution.
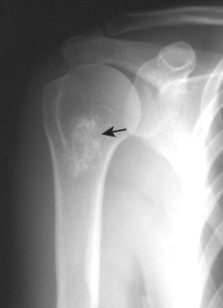 |
| FIG. 13-46 An irregular pattern of medullary calcification appearing in the proximal humerus (arrow) of an asymptomatic middle-aged adult. The presentation is consistent with an enchondroma. Other consideration should be given to intramedullary bone infarct or central chondrosarcoma. The latter usually is accompanied by pain. |
The presence of multiple enchondromas (enchondromatosis, or Ollier’s disease) increases the risk of malignant transformation by 30%. 16,44,211 The combination of multiple enchondromas and subcutaneous hemangiomas is termed Maffucci’s syndrome and is associated with a 30% or greater increased risk for malignant transformation than occurs with solitary enchondromas. 4,121,232 Malignant transformation is usually to a chondrosarcoma.
Juxtacortical chondromas are found in patients predominantly before the age of 30 years, 23 appearing more commonly in males than females, at a ratio of 2:1.
IMAGING FINDINGS
Chondromas, whether medullary or juxtacortical, solitary or multiple, usually are found in the small bones of the hands, particularly the proximal phalanges (Figs. 13-47 and 13-48), and less commonly the feet (Fig. 13-49). The humerus, femur, and tibia also are involved.
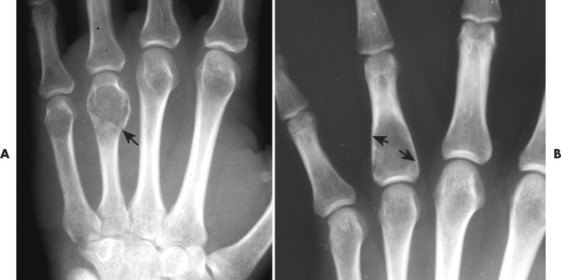 |
| FIG. 13-47 Enchondroma. A, Fractured enchondroma of the hand (arrow). Enchondromas of the hand commonly appear as slightly expansile, geographic lesions, usually with a well-defined pattern of matrix calcification. B, Faint, mildly expansile cystic defect noted in the proximal aspect of the proximal phalanx of the fourth digit (arrows). These lesions do not exhibit prominent matrix calcification that is common for enchondromas in this location. |
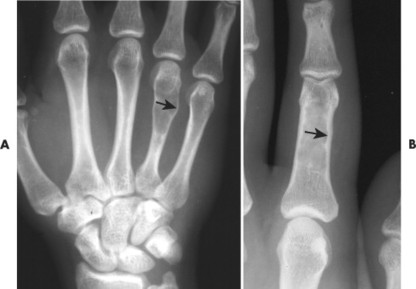 |
| FIG. 13-48 A, Oblong enchondroma with scalloping of the inner cortex of the lesion (arrow), a characteristic feature, in a 12-year-old boy. B, Faint lesion in a second patient demonstrates similar endosteal scalloping. (B, Courtesy Robert C. Tatum, Davenport, IA.) |
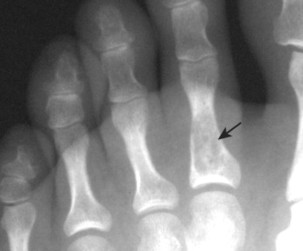 |
| FIG. 13-49 Small cystic defect of the proximal phalanx of the foot’s second digit (arrow). This lesion exhibits a faint punctate pattern of matrix calcification consistent with the cartilage origin of the lesion. (Courtesy Jay Brammier, Durant, IA.) |
An enchondroma appears as a well-defined medullary lesion, most often with some degree of calcification, endosteal scalloping (see Fig. 13-48), and bone expansion. Enchondromas are usually small, less than 3 cm in diameter (Fig. 13-50). On MRI the calcific foci appear as minute signal voids (Fig. 13-51). Enchondromas often are not as clearly outlined in long bones as when they appear in small tubular bones.
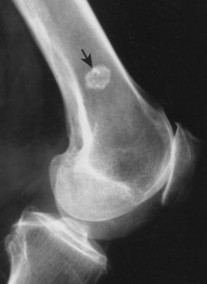 |
| FIG. 13-50 Calcific enchondroma of the distal femur (arrow). (Courtesy Jack Avalos, Davenport, IA.) |
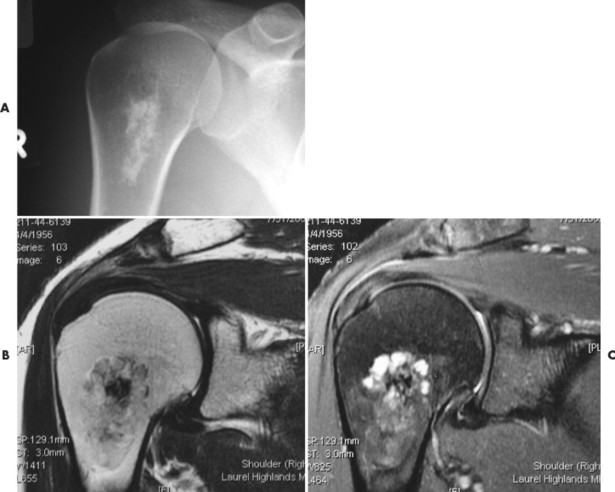 |
| FIG. 13-51 Enchondroma appearing on, A, plain film. B, T1- and, C, T2-weighted magnetic resonance imaging of the proximal humerus. The lesion is well circumscribed radiographically. The T2-weighted coronal image demonstrates small lobulated foci of increased signal intensity separated by a collagen mesh of decreased signal intensity. (Courtesy John Rizzo, Edensburg, PA.) |
Malignant transformation to a chondrosarcoma is a rare event, occurring in less than 1% of patients. Malignant transformation occurs more often in proximal lesions (e.g., humerus versus phalanx), and is suggested by the disappearance of a previously noted pattern of matrix calcification.
Juxtacortical chondromas appear as soft-tissue masses with erosion or saucerization of the adjacent cortex (Figs. 13-52 and 13-53). Calcification is noted in 50% of juxtacortical chondromas. 240
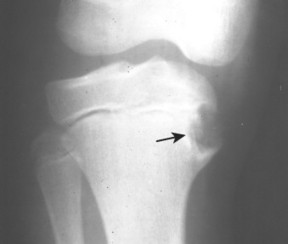 |
| FIG. 13-52 Periosteal chondroma (arrow) of the medial portion of the proximal tibia. (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
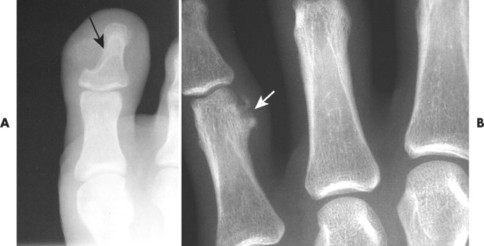 |
| FIG. 13-53 A, Patient exhibiting periosteal chondroma of the distal phalanx of the first pedal digit (arrow). B, Second patient exhibiting involvement in the proximal phalanx of the hand (arrow). (A, Courtesy Ian D. McLean, LeClaire, IA.) |
Ollier’s disease appears with multiple (usually many) cystic defects of the hands, and less commonly the feet (FIG. 13-54FIG. 13-55FIG. 13-56 and FIG. 13-57). Soft-tissue hemangiomas are signified radiographically as soft-tissue masses with multiple phleboliths (Fig. 13-58).
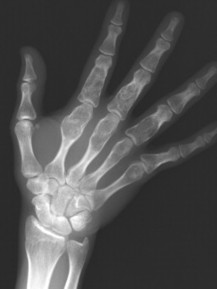 |
| FIG. 13-54 Ollier’s disease, defined as multiple enchondromas. Each lesion presents as a slightly expansile cystic defect with well-defined central matrix calcification. (Courtesy Gary Longmuir, Phoenix, AZ.) |
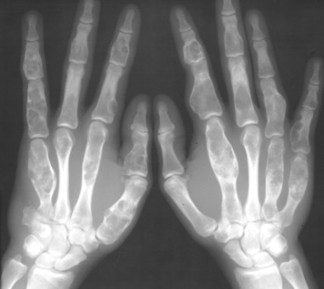 |
| FIG. 13-55 Ollier’s disease. A 28-year-old male patient with a bilateral presentation of multiple enchondromas throughout the small bones of the hands. A patient with Ollier’s disease is at greater risk of experiencing malignant transformation to a chondrosarcoma than is someone with a solitary enchondroma presentation. |
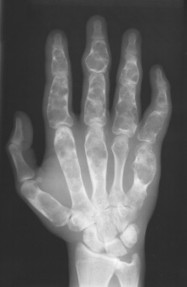 |
| FIG. 13-56 Ollier’s disease defined by the multiple enchondromas, that in this case appear symmetrically placed within the small bones of the hands. (Courtesy Beverly Harger, Portland, OR.) |
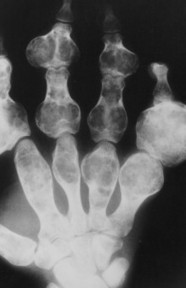 |
| FIG. 13-57 Ollier’s disease exhibiting individual lesion with marked expansile appearance and characteristic stippled matrix calcification. At quick glance, the appearance is reminiscent of gout, except the former has interosseous lesions, the latter soft-tissue tophi. (Courtesy Joseph W. Howe, Sylmar, CA.) |
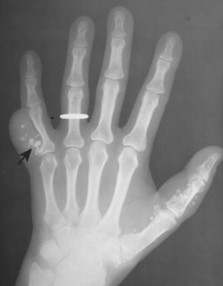 |
| FIG. 13-58 Maffucci’s syndrome describes the concurrent presentation of multiple enchondromas (Ollier’s disease) and soft-tissue hemangiomas within (arrow). (Courtesy Steven P. Brownstein, MD, Springfield, NJ.) |
CLINICAL COMMENTS
Chondromas typically are asymptomatic. The presence of associated pain increases the clinical suspicion for the presence of pathologic fracture, malignant transformation, or a low-grade de novo chondrosarcoma. In addition, any chondroma that exhibits continued growth in a skeletally mature patient should be closely scrutinized to exclude malignancy because lesions typically stop growing in adult patients. Surgical curettage usually is indicated for substantial lesions.
• Chondromas are the most common tumor of the phalanges.
• Chondromas have a medullary (enchondroma) or juxtacortical (perichondroma) location.
• They usually occur in patients younger than age 30 years.
• Matrix calcification occurs in 50% of patients with chondromas.
• Malignant transformation to a chondrosarcoma is a rare occurrence (1% of patients) unless multiple lesions are present (30% of patients).
Chondroblastoma
Related posts:
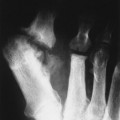
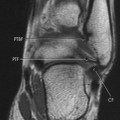
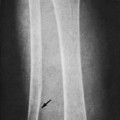
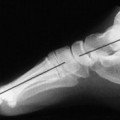
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


