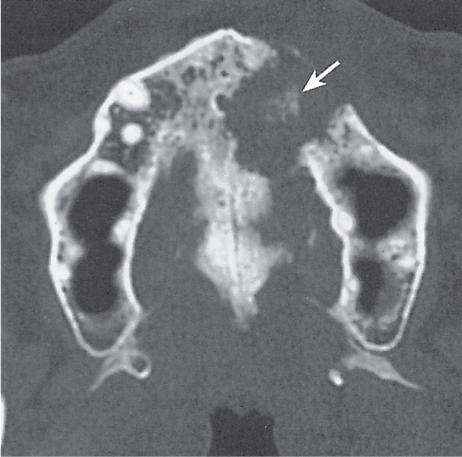
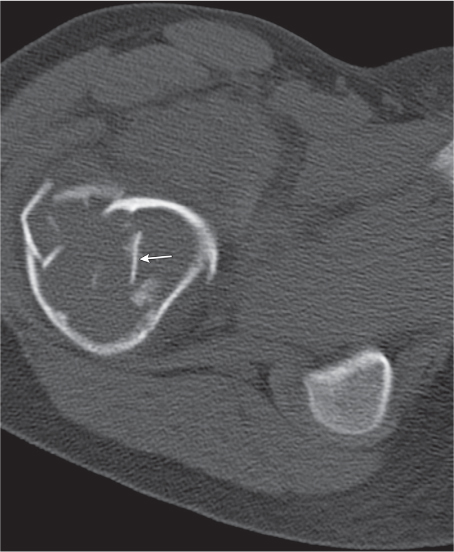
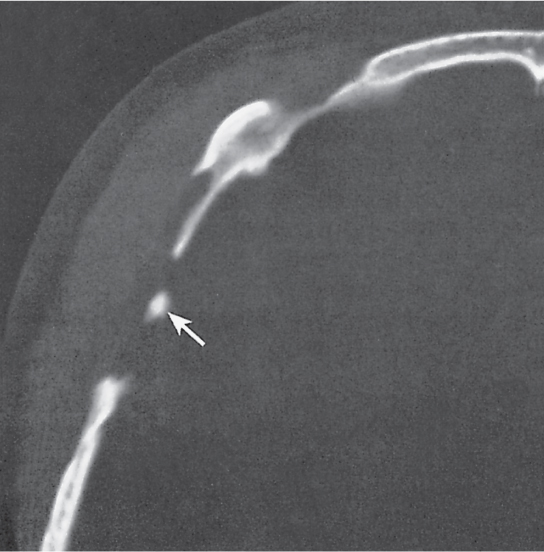
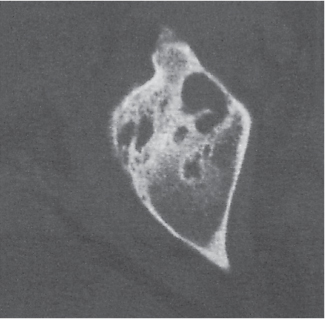
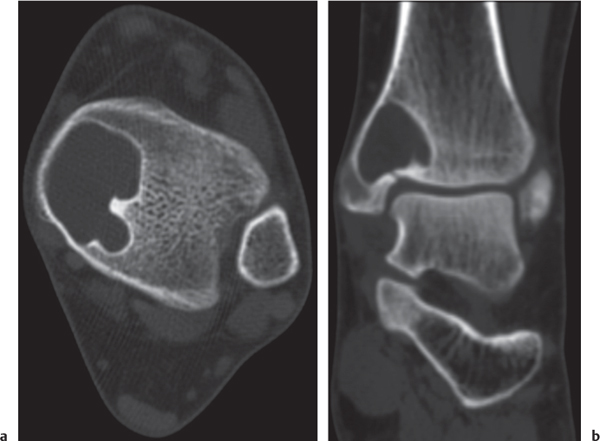
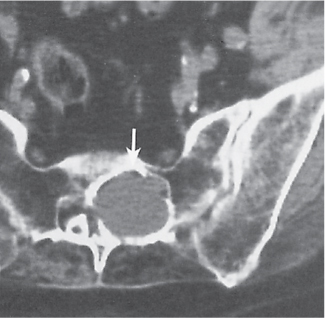
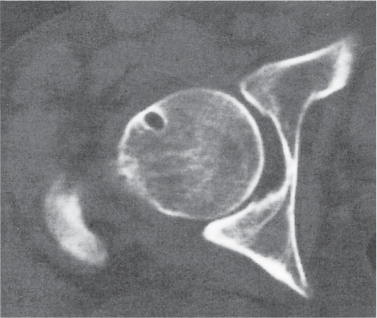
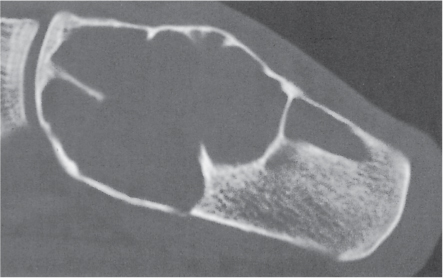
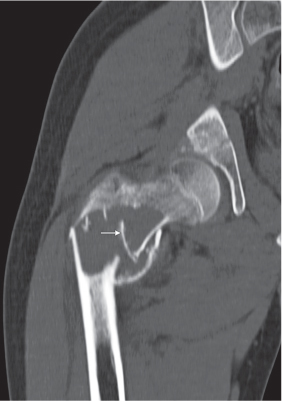
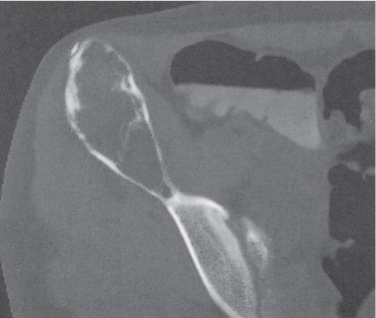
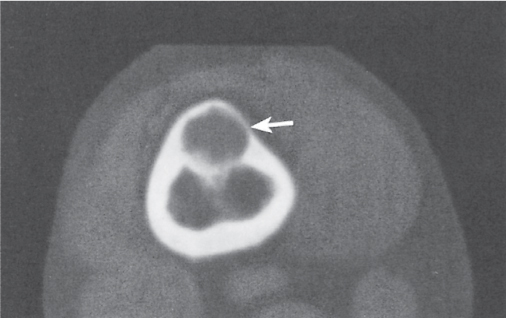
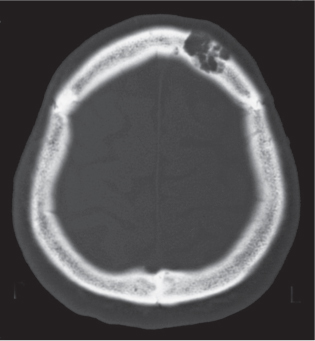
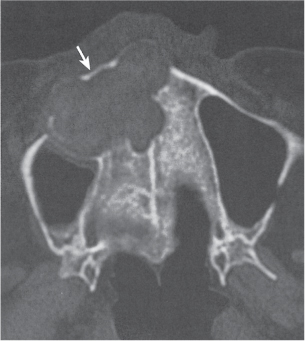
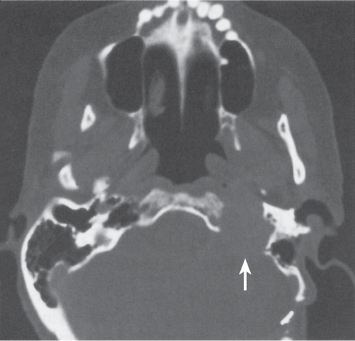
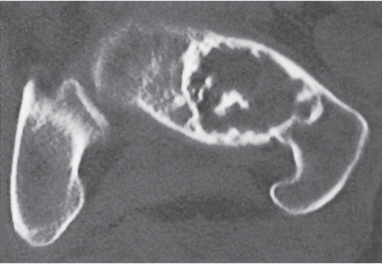
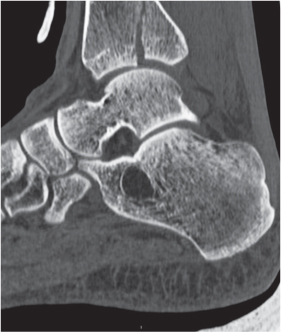
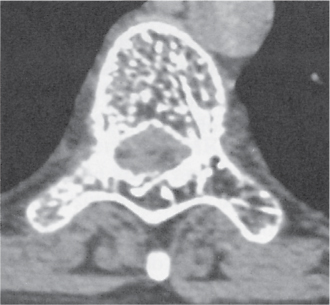
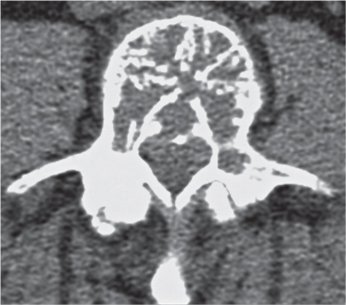
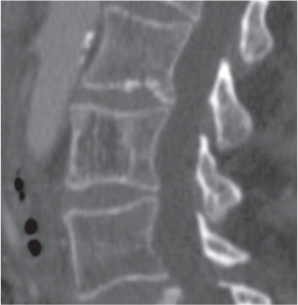
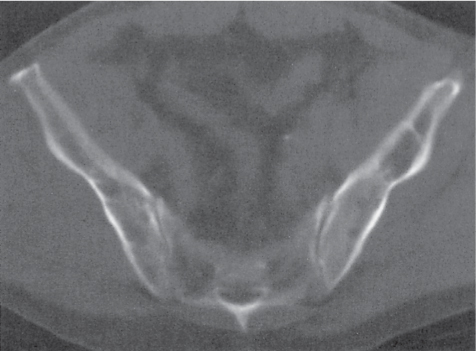
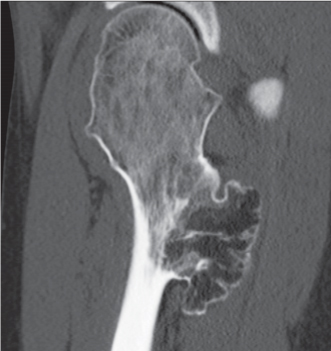
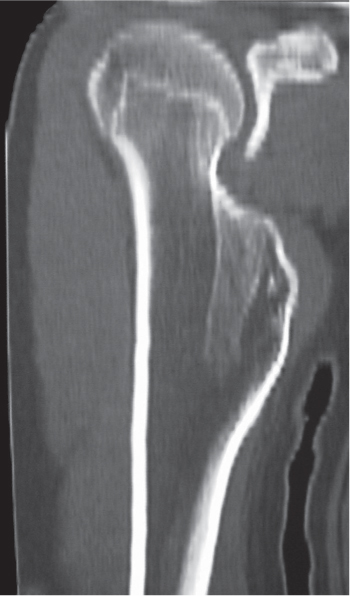
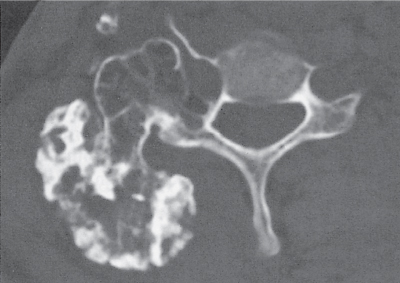
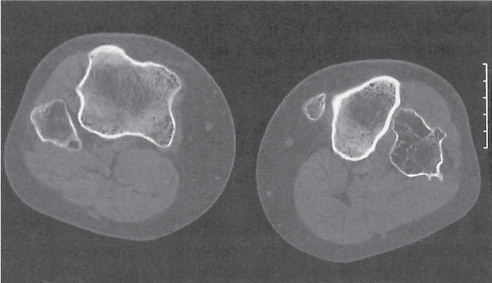
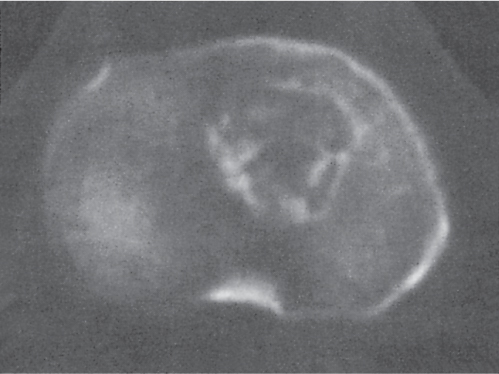
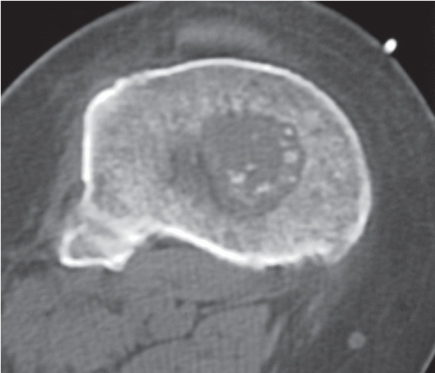
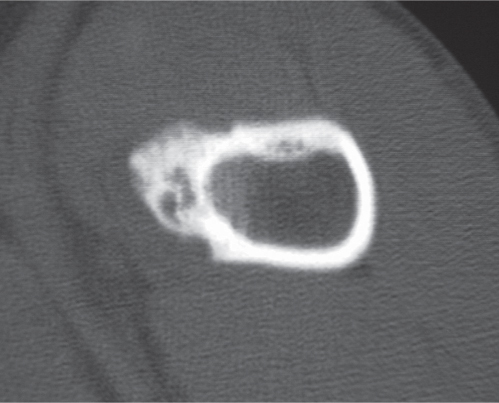
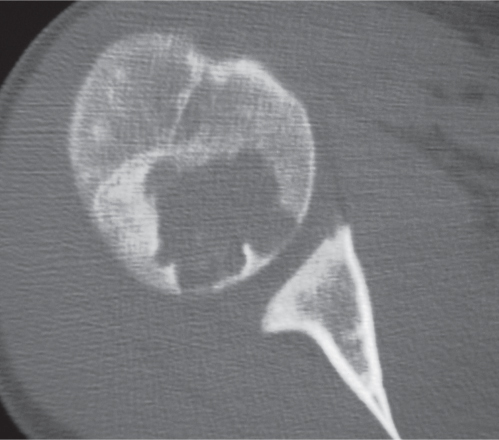
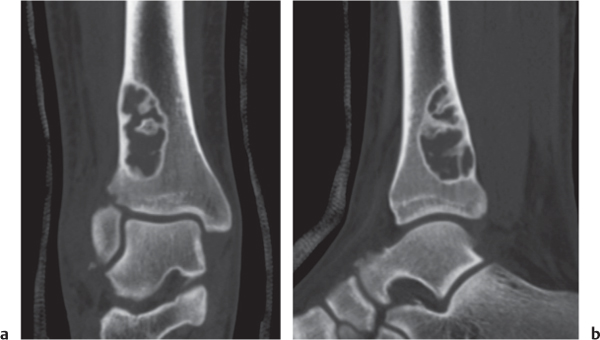
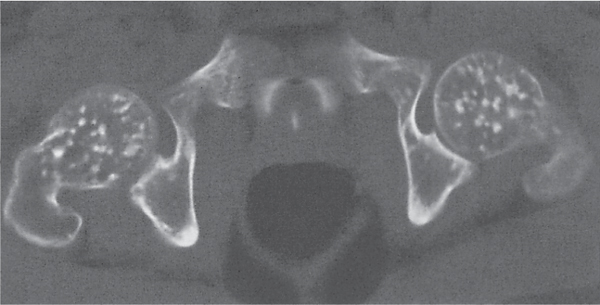
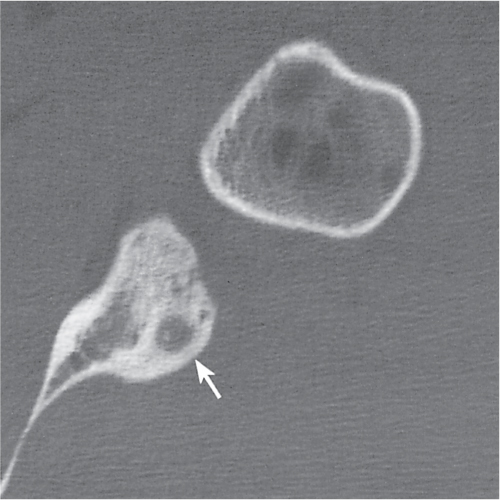
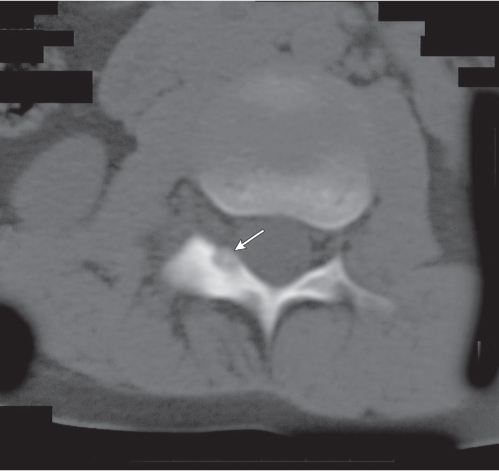
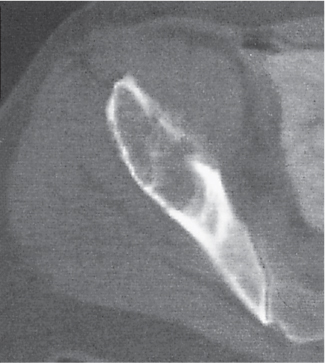
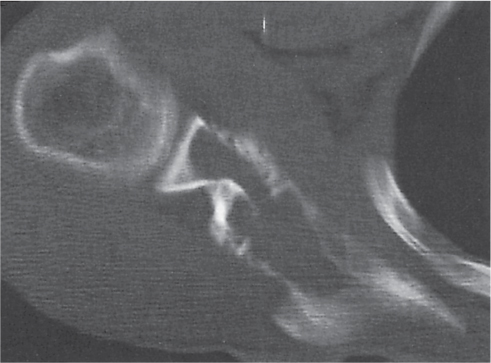
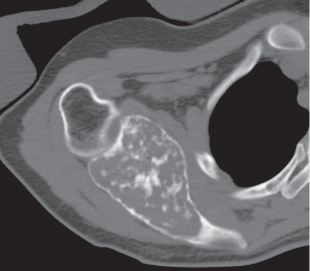
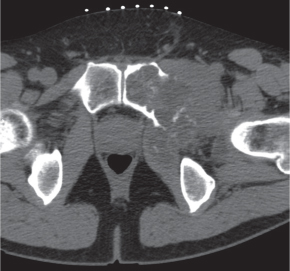
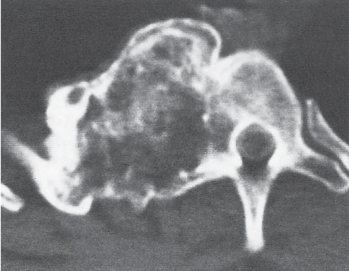
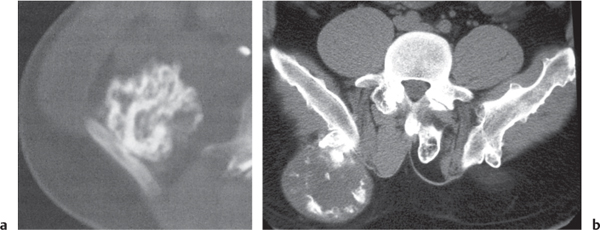
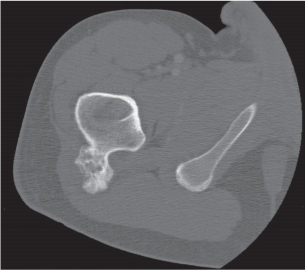
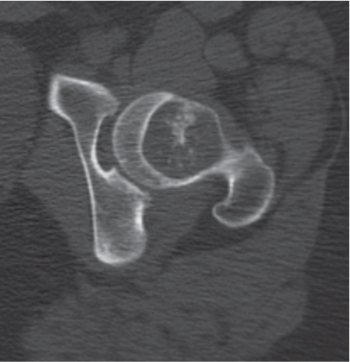
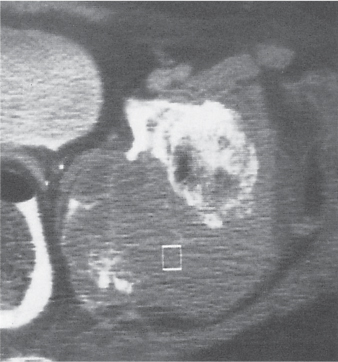
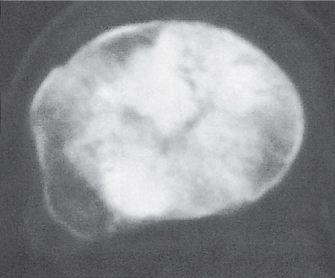
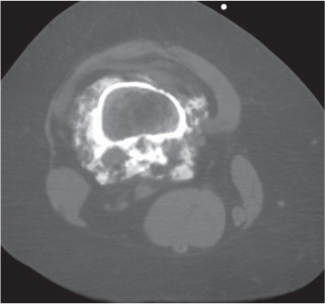
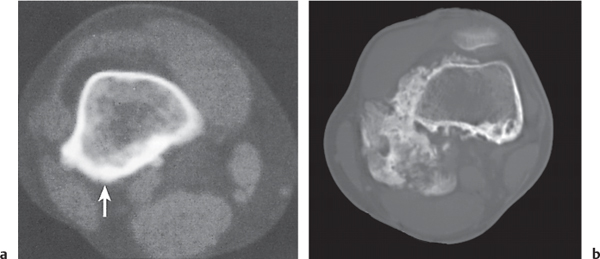
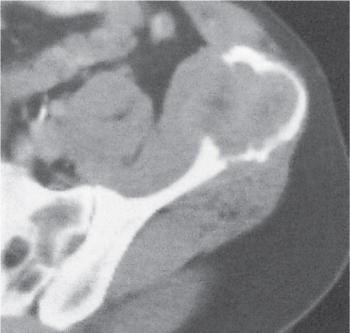
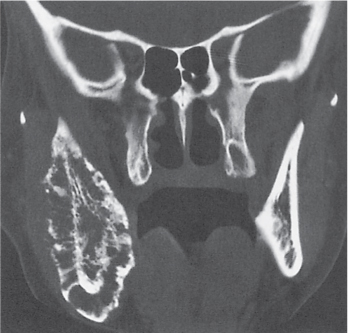
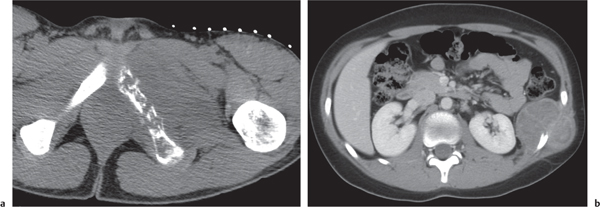
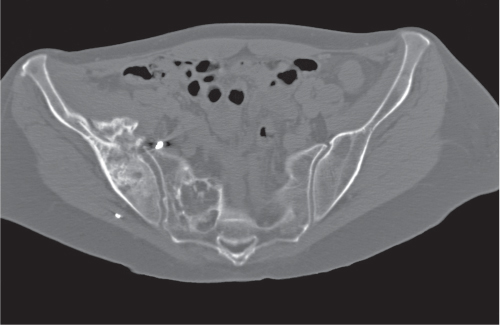
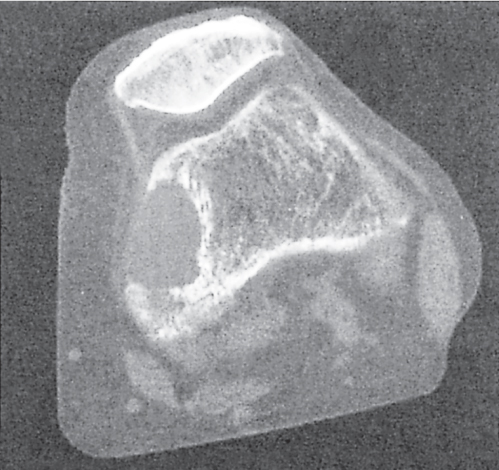
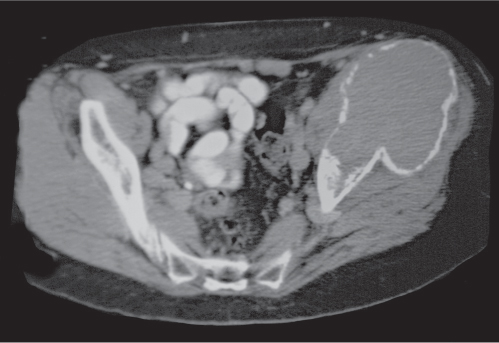
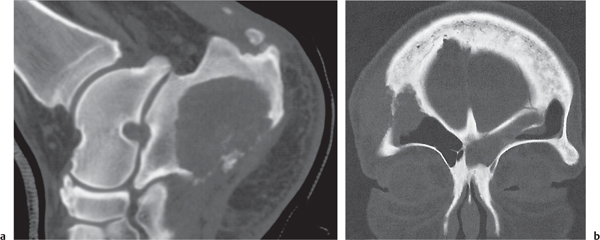
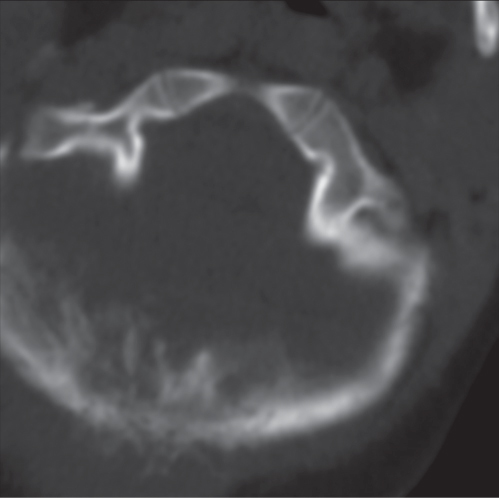
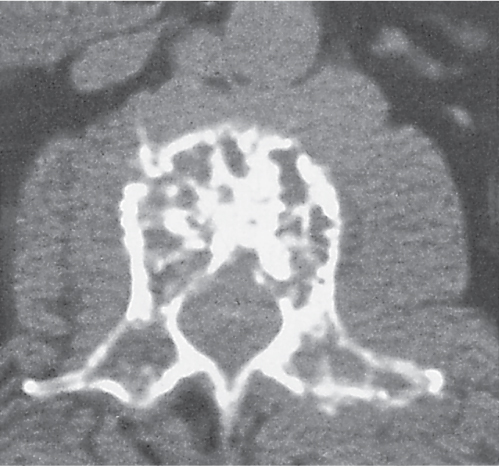
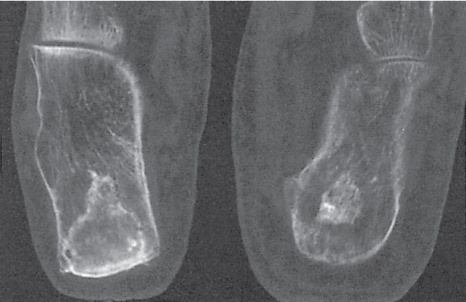
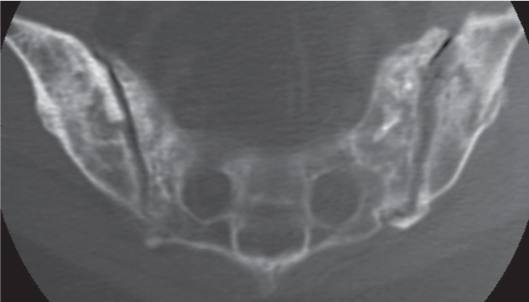
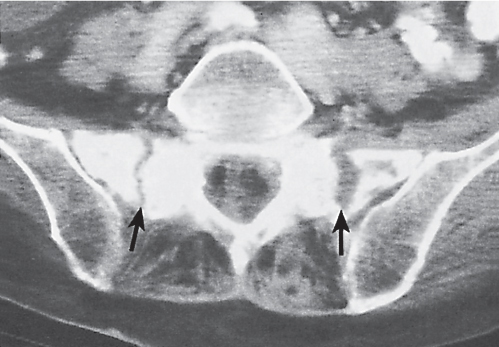
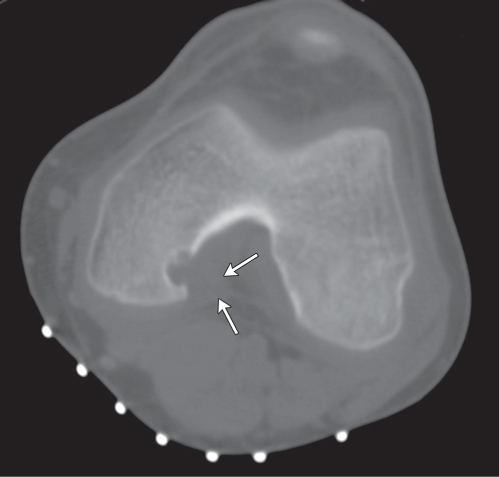
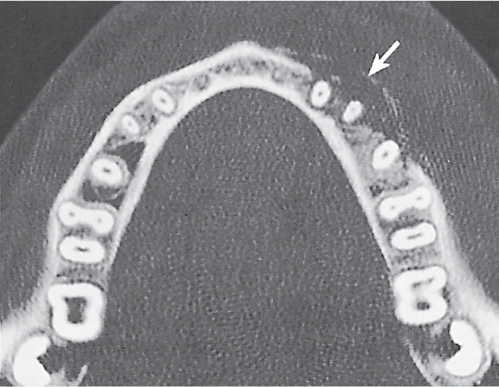
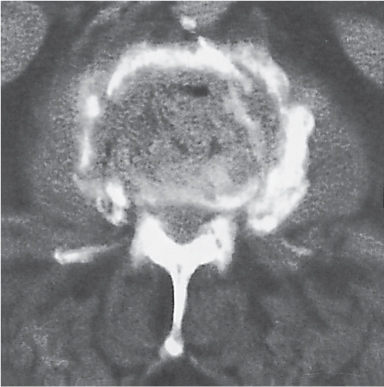
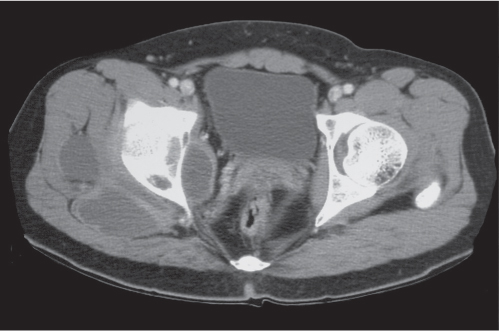
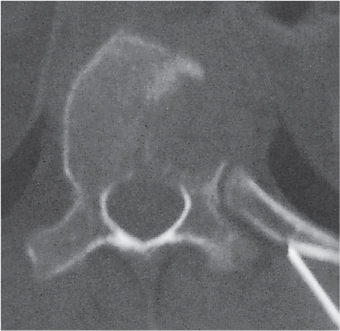
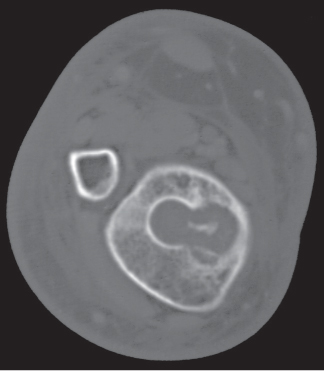
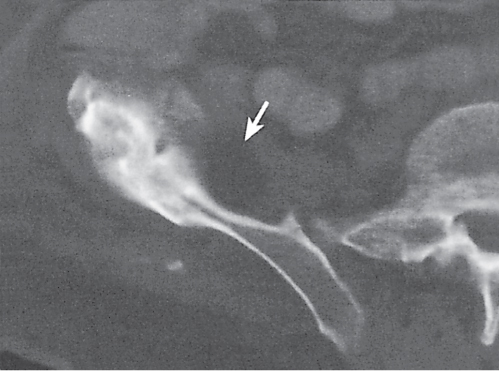
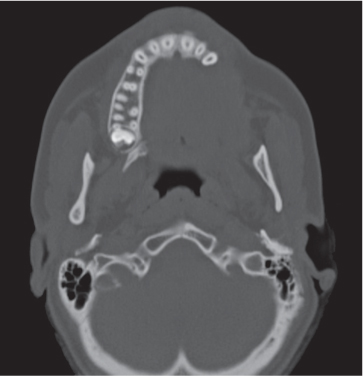
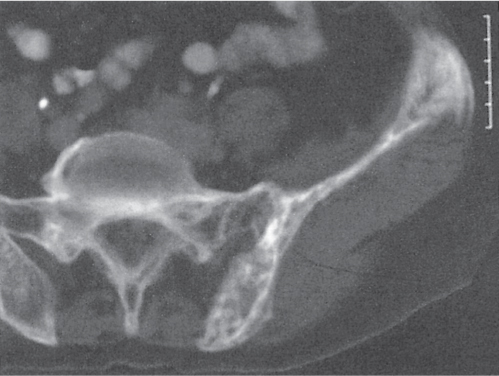
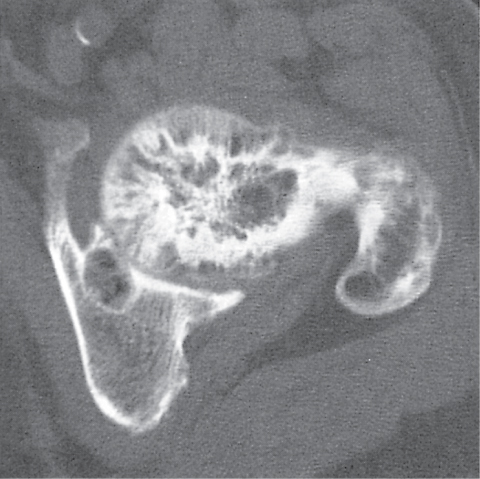
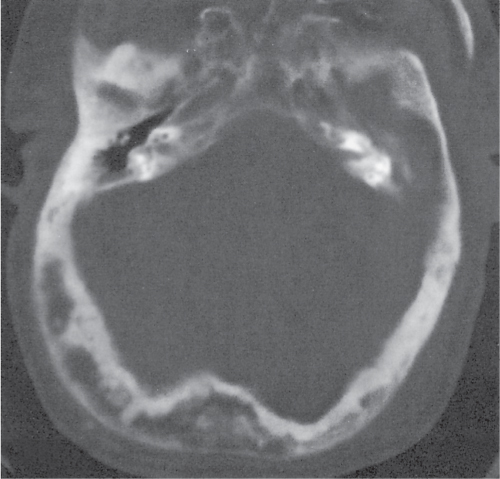
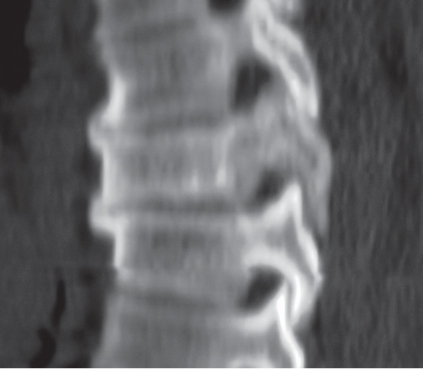
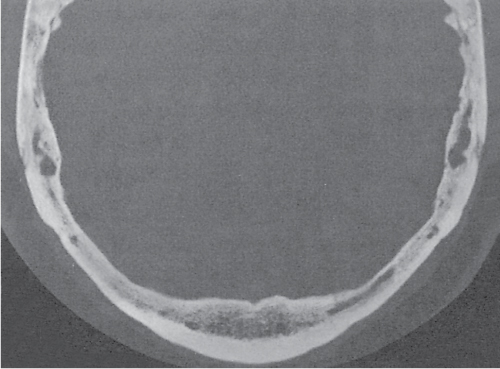
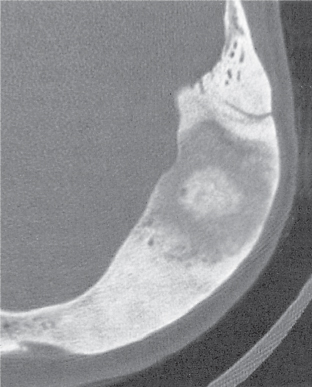
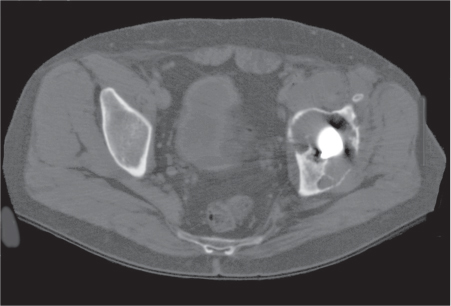
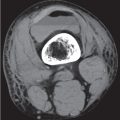
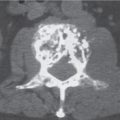
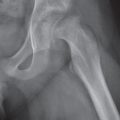
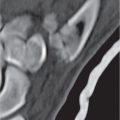
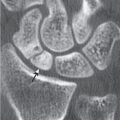
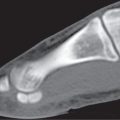
Subchondral cyst (geode)
Fig. 14.8 |
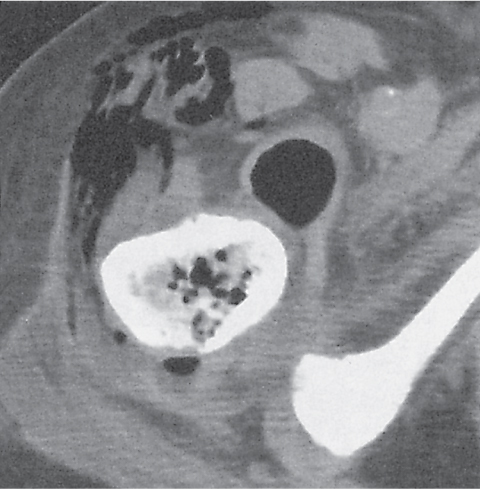
Solitary or multiple lytic defects measuring up to 3 cm in diameter with typically a thin sclerotic border. Communication with the adjacent arthritic joint is often identifiable. Gas is occasionally found in these lesions (pneumatocysts), especially about the sacroiliac joints. |
Associated with osteoarthritis (primary and posttraumatic), rheumatoid arthritis, avascular necrosis, and calcium pyrophosphate deposition disease (CPDD). Differential diagnosis: An intraosseous tophus in chronic gouty arthritis that may or may not be calcified must be differentiated. |
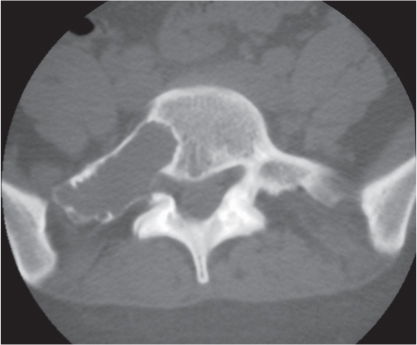
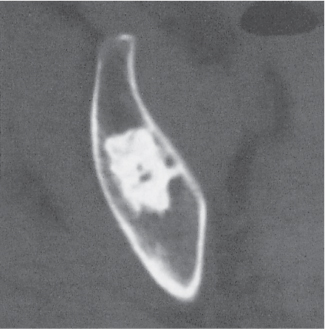
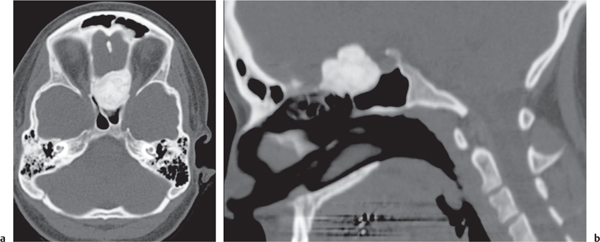
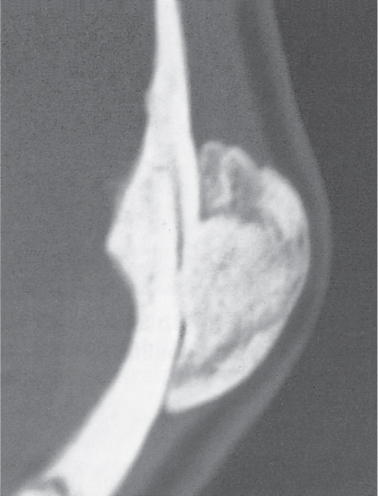
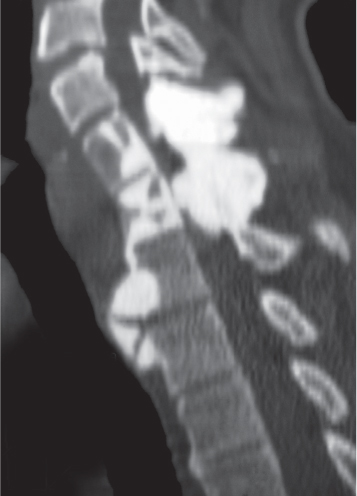
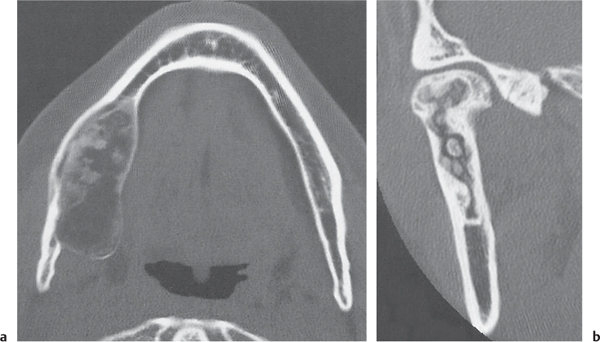
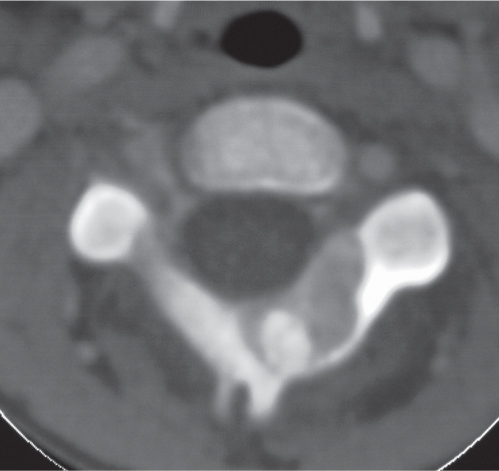
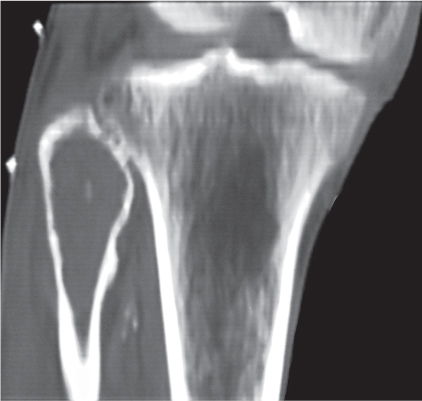
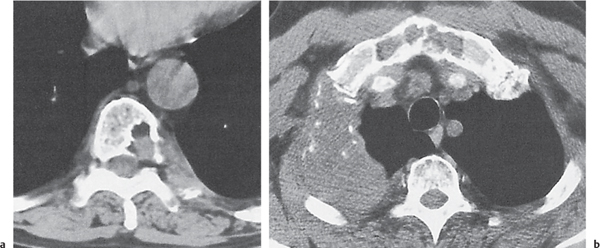
Intraosseous ganglion
Fig. 14.9a, b
Fig. 14.10 |
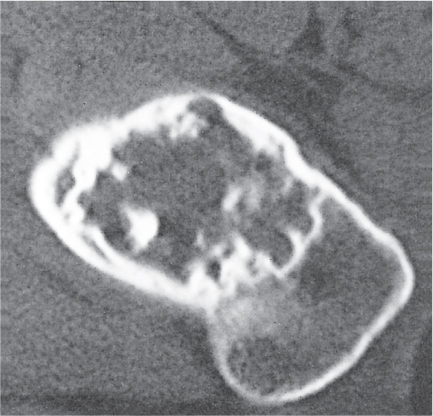
Unilocular or, less commonly, multilocular, well-defined lytic epiphyseal lesion, often with a sclerotic margin. It may be associated with an extraosseous component of near-water to soft tissue density (soft tissue ganglion). Preferred locations are the medial malleolus of the tibia, femoral head, acetabulum, and carpal bones. Characteristically, the lesion does not communicate with the adjacent joint and is not associated with an arthritic process. |
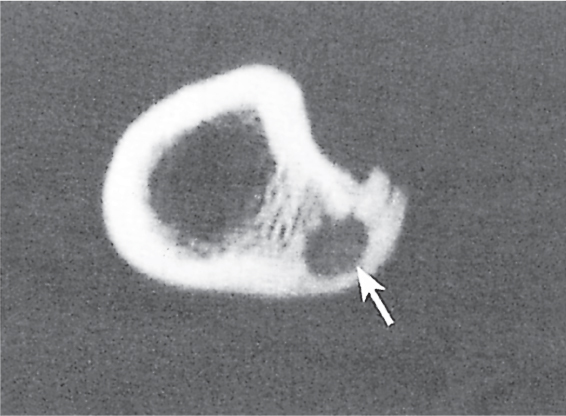
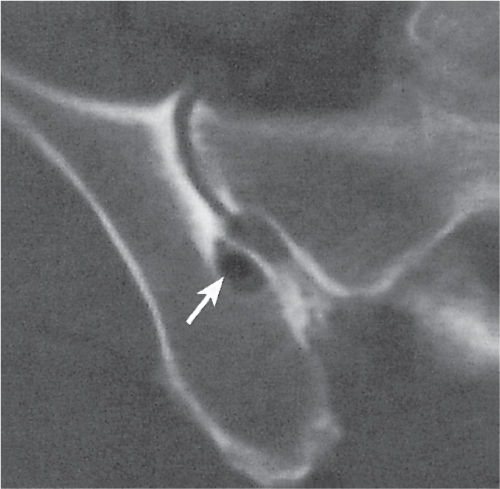
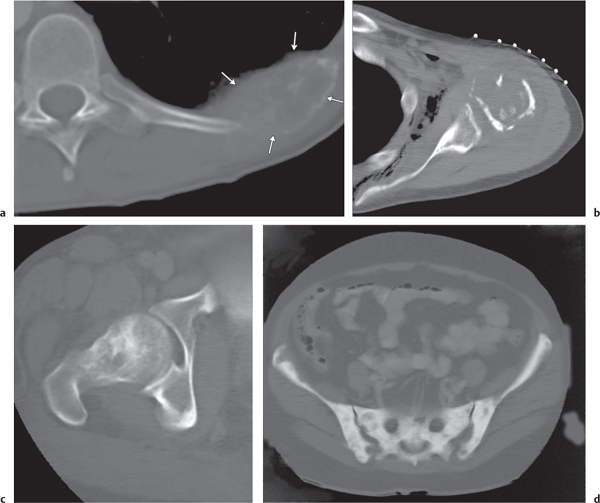
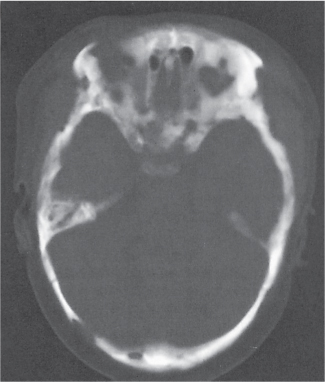
A herniation pit (Fig. 14.11) is a well-circumscribed radiolucent defect usually measuring < 1 cm in diameter with a thin sclerotic border in the anterolateral aspect of the proximal femoral neck, which may represent an incidental finding or is associated with femoroacetabular impingement (FAI). |
Echinococcosis |
Complex, expansile, trabeculated, osteolytic lesion (s). Cortical violation and adjacent soft tissue mass formation, often with calcification, may be associated. Vertebral column, pelvis, long bones, adjacent ribs, and skull are most frequently involved. |
Caused by Echinococcus granulosus (hydatid disease) or, less commonly, Echinococcus multilocularis. Cysts may develop in various viscera, particularly the liver and the lungs. Calcification of the cyst wall is common except in the lung. |
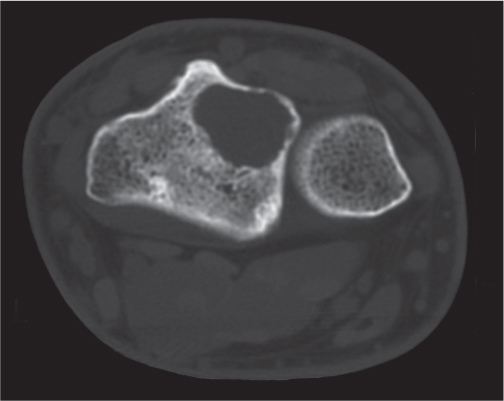
Simple (unicameral) bone cysts
Fig. 14.12
Fig. 14.13 |
Centrally located, slightly expansile osteolytic lesion with cortical thinning. A central component of near-water density is characteristic but not always appreciated. Preferred locations in children are the metaphyses of long tubular bones (especially humerus and femur) and in adults the calcaneus and the ilium of the pelvis. Diaphyseal cysts in long bones are frequently large, expansile, and multiloculated. Fluid levels may be present. |
In long tubular bones of children, the metaphyseal cyst is juxtaposed to the physis and frequently has an elongated shape paralleling the bone axis. A pathologic fracture is common. The fracture fragment within the cyst drops to the dependent portion of the lesion (“fallen fragment” sign).
A pneumatocyst is diagnosed in the presence of either intralesional gas or a gas–fluid level. |
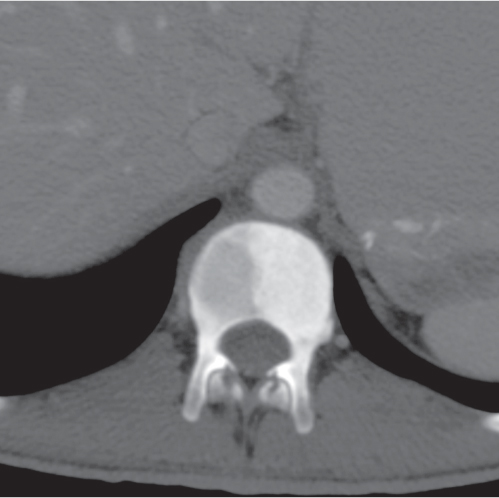
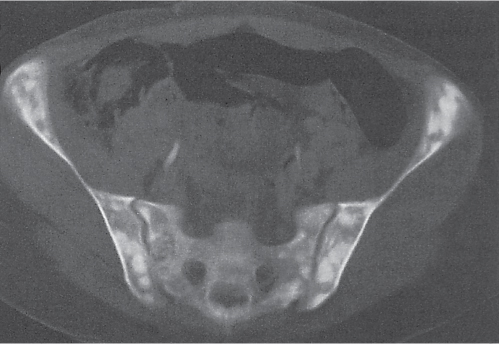
Aneurysmal bone cyst
Fig. 14.14
Fig. 14.15 |
Presents most commonly either as an eccentric, expansile, osteolytic lesion with thin cortical shell or with extraosseous soft tissue component located in the metaphysis of a long tubular bone. Sclerosis at the site where the periost is lifted by the lesion (“buttressing”) is common. Fine trabeculation and fluid levels within the lesion are characteristic. Other common locations are the posterior elements of the spine and the innominate bone, where a large soft tissue component may simulate a malignancy. |
Primary lesions are found in 80% of patients younger than age 20 y.
Secondary aneurysmal bone cysts arise in preexisting bone lesions, such as giant cell tumors, osteoblastomas, chondroblastomas, telangiectatic osteosarcomas, malignant fibrous histiocytomas, fibrous dysplasia, and Paget disease. |
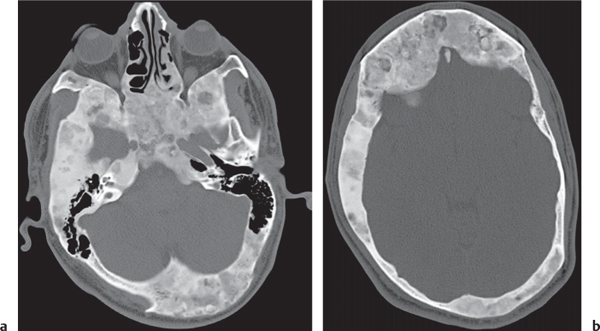
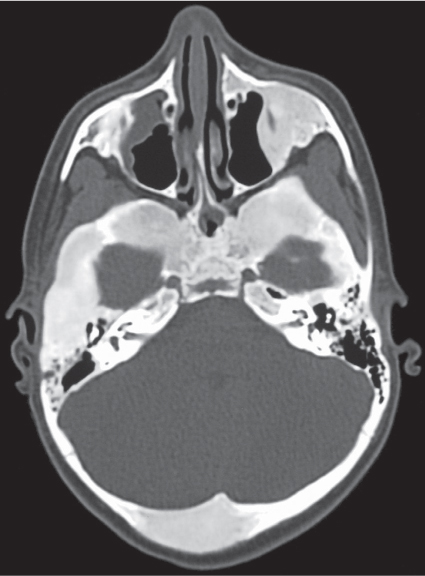
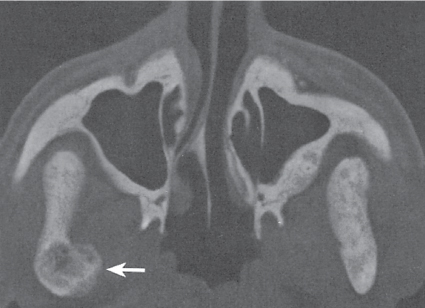
Epidermoid (inclusion cyst)
Fig. 14.16
Fig. 14.17 |
Intraosseous lesions present as well-defined osteolytic defects often with thin sclerotic margin occurring in the terminal phalanges of the hand and in the skull. Lesions may be expansile with thinning or destruction of the cortex. The cyst content consists of thick, waxy material rich in cholesterol varying from −5 to 20 HU. A button sequestrum is frequently seen in calvarial lesions. |
Usually observed in the second to fourth decades of life. A history of trauma is frequently present, suggesting intraosseous implantation of ectodermal tissue with subsequent development of an epidermoid. The cysts are lined with a stratified squamous epithelium shedding keratin debris that breaks down and forms cholesterol. |
Glomus tumor
Fig. 14.18 |
Osteolytic lesions with marked contrast enhancement occurring in the terminal phalanges of the hand and temporal bone. |
In the fingertips, the lesion is indistinguishable on plain radiographs from an epidermoid (inclusion cyst). |
Lipoma
Fig. 14.19
Fig. 14.20 |
Well-circumscribed osteolytic lesion, often surrounded by a sclerotic border. May contain a central calcified nidus (especially in the calcaneus). Low attenuation of the fatty tissue is virtually diagnostic. |
Rarely, fatty degeneration secondary to infarction and lesion containing histiocytes with fat vacuoles may also demonstrate negative CT values.
Liposclerosing myxofibroma is a rare benign, well-defined lytic lesion typically surrounded by a sclerotic border in the pretrochanteric or intertrochanteric region of the proximal femur. However, fat is not invariably demonstrated by CT.
Liposarcomas rarely arise in bone. |
Hemangioma
Fig. 14.21
Fig. 14.22
Fig. 14.23 |
Spine: Small focal osteolytic lesion with trabeculae often arranged in a honeycomb or “cartwheel” configuration to coarse, vertical trabecular pattern (“corduroy” appearance) involving the vertebral body. The diameter of thickened vertical trabeculae is rather uniform. (Differential diagnosis: Multiple myeloma, metastases, and Paget disease, which may present with irregular, coarse trabecular thickening.) Extension into the posterior elements, paraspinal soft tissues, and spinal canal occurs but is not common.
Skull: Slightly expansile osteolytic lesion with lattice-like (“cartwheel”) or radiating (“sunburst”) pattern.
Pelvis and tubular bones: Poorly defined osteolytic lesion with lattice-like trabecular pattern. Involvement is uncommon. |
Usually found in patients older than 40 y, with female predominance. Occasionally non–Hodgkin lymphoma (especially the histiocytic type) and metastases (e.g., breast carcinoma) may simulate a hemangioma in the spine.
Cystic angiomatosis (Fig. 14.24) is characterized by widespread cystic bone lesions that are frequently combined with visceral involvement. |
Osteochondroma
Fig. 14.25
Fig. 14.26
Fig. 14.27 |
Bony protuberance demonstrating cortical and medullary contiguity with parent bone is diagnostic in tubular bones. The lesion may be pedunculated (with narrow stalk and bulbous tip) or sessile (with broad, flat base). Osteochondromas characteristically originate from the metaphyses and point away from the nearby articulation. The tip of the osteochondroma is covered by a hyaline cartilage cap that may contain regular stippled calcifications. A large cartilaginous cap thicker than 2 cm often with irregular calcifications is suspicious for malignant transformation. The thickness of the cartilage cap is best assessed with MRI.
In the pelvis, osteochondromas are frequently large and difficult to differentiate from lesions that have undergone malignant transformation.
In the ribs, osteochondromas frequently occur at the costochondral junction. |
Osteochondromas occur in children and adolescents as a slow-growing, painless mass. They are intimately related to the physis and cease to enlarge with fusion of the adjacent growth plates. Rarely osteochondromas may develop in adults after trauma.
Differential diagnosis:
1. Supracondylar process of the humerus: Spur originating from the anteromedial aspect of the distal humerus pointing toward the elbow (phylogenetic vestige).
2. Pes anserinus spur in the medial aspect of the proximal tibia (enthesophyte, occasionally associated with anserinus bursitis).
3. Posttraumatic spurs secondary to healed avulsion fractures.
4. “Tug” lesions at tendinous attachment sites in the lower extremity.
5. See also periosteal (juxtacortical) chondroma and bizarre parosteal osteochondromatous proliferation (BPOP) in this table.
Hereditary multiple exostoses (Fig. 14.28) present with multiple, usually bilateral, and symmetric osteochondromas in the axial and appendicular skeleton associated with bone modeling deformities. |
Dysplasia epiphysealis hemimelica (Trevor disease) |
Lobulated osseous mass (articular chondroma) originating in an epiphysis, carpal, or tarsal bone. Presents initially in infants with irregular calcifications/ossifications on one side of an enlarged epiphysis or a carpal/tarsal bone. Preferential involvement is the medial side of a lower extremity epiphysis (e.g., distal femur, proximal and distal tibia) or the medial side of a tarsal bone (e.g., talus). Multiple bones in a single extremity are affected in two thirds of cases. |
Presents in children and young adults with swelling, pain, and deformity localized to one side of the body. Histologically, the pedunculated mass with cartilaginous cap is indistinguishable from an osteochondroma. |
Enchondroma
Fig. 14.29
Fig. 14.30 |
Well-circumscribed lesion, often with endosteal scalloping composed of hyaline-type cartilage with varying degrees of calcifications. Preferred locations are the metaphyses of the long tubular bones and the diaphyses in the short tubular bones of the hands and feet. In the long tubular bones, larger lesions with sizable areas of uncalcified matrix should suggest the possibility of malignant transformation or a low-grade chondrosarcoma. Other imaging features in long tubular bones suggesting low-grade malignancy include extensive endosteal scalloping (more than two thirds of the cortical thickness or more than two thirds of the length of the lesion) and solid periosteal reaction/localized cortical thickening about the lesion. Frank cortical destruction and associated soft tissue mass are virtually diagnostic for a chondrosarcoma. |
Enchondromas are usually discovered in the third or fourth decade of life as incidental findings or painless swelling. The presence of pain should arouse suspicion of malignant transformation.
Enchondromatosis (Ollier disease) is characterized by multiple, asymmetrically distributed enchondromas often in deformed tubular bones and the pelvis. In Maffucci syndrome, multiple soft tissue cavernous hemangiomas with phleboliths are associated with enchondromatosis with predilection for tubular bones of the hands and feet. |
Periosteal (juxtacortical) chondroma
Fig. 14.31 |
Soft tissue mass with erosion of the adjacent cortex and varying degrees of periosteal new bone formation, including buttressing (thickening of the cortex at the distal and proximal margins of the lesion) is typical. Matrix calcification is evident in the majority of cases and may be extensive. Metaphyses of the long tubular bones and hands are most commonly affected. |
All ages are affected, but usually diagnosed under the age of 30 y. Slight male predominance. |
Bizarre parosteal osteochondromatous proliferation (BPOP) |
Well-marginated sessile or pedunculated mass of heterotopic ossification arising from the cortical surface without medullary contiguity between lesion and adjacent bone is characteristic. |
Occurs usually in the hands and feet (occasionally in long tubular bones) without age and gender predilection. A history of trauma is evident in some patients.
Florid reactive periostitis involving most commonly the proximal or middle phalanges of the hands is best considered a variant of traumatic myositis ossificans. |
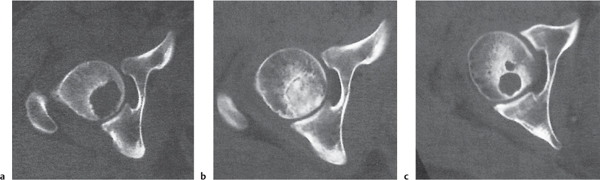
Chondroblastoma
Fig. 14.32 |
Well-defined lytic epiphyseal or apophyseal lesion with or without a sclerotic border and matrix calcifications. A solid periosteal reaction in the adjacent metaphyseal shaft may be induced by bone marrow edema. Approximately 10% of chondroblastomas occur in the hands and feet with predilection for the talus and calcaneus. |
Uncommon benign cartilaginous lesion occurring between the ages of 5 and 25 y with slight male predominance. |
Chondromyxoid fibroma |
Eccentric metaphyseal osteolytic lesion with cortical expansion, coarse trabeculation, endosteal scalloping, and scalloped sclerotic medullary border is a typical presentation. Destruction of the cortex resulting in a hemispherical osseous defect or “bite” without periosteal reaction is a characteristic finding in larger lesions. Predilection for the long tubular bones, especially of the lower extremity, but occasionally also found in the pelvis, foot, and hand. |
Uncommon benign cartilaginous lesion occurring between the ages of 5 and 25 y with slight male predominance. Slowly progressive pain, tenderness, swelling, and restriction of motion are typical.
Fibrocartilaginous mesenchymoma is a very rare benign solitary expansile osteolytic lesion with spotty or ringlike calcifications, cortical destruction, and soft tissue invasion originating most often in the metaphysis of a long tubular bone. Age distribution is similar to chondromyxoid fibroma. |
Periosteal (juxtacortical) desmoid |
Saucer-like defect in the posteromedial cortex of the distal femur, often associated with adjacent sclerosis, periostitis, and soft tissue swelling. |
Occurs between the ages of 15 and 20 y. Considered to be a sequela of trauma at the insertion of the adductor magnus muscle. |
Nonossifying fibroma
Fig. 14.33a, b |
Small lesions present as round or oblong, well-delineated, radiolucent areas in the cortex with normal or sclerotic adjacent bone. They typically arise in the metaphysis at a short distance from the physis. Larger lesions present as a well-delineated, eccentric, oblong osteolytic area in the diametaphysis. They frequently have a multiloculated appearance, and both cortical expansion and thinning may be evident. The long tubular bones, especially the tibia and femur, are most frequently affected. With time the lesions may spontaneously disappear or become sclerotic. |
Usually diagnosed in patients younger than 20 y. Smaller lesions are referred to as benign fibrous cortical defects. They are asymptomatic and diagnosed as an incidental finding on routine radiography and occasionally are multifocal. They usually regress spontaneously or less commonly enlarge and migrate with growth into the diaphyses, eventually being referred to as nonossifying fibromas. Pathologic fractures in larger lesions are not uncommon.
Jaffe–Campanacci syndrome consists of multiple nonossifying fibromas associated with café-au-lait spots, mental retardation, and hypogonadism.
Benign fibrous histiocytomas (fibroxanthoma) are histologically identical to nonossifying fibromas but present as slightly more aggressive lesions in patients older than 20 y without site predilection. |
Desmoplastic fibroma
Fig. 14.34 |
Central, often trabeculated (soap bubble or honeycomb pattern) osteolytic lesion in the metaphyses of long tubular bones, mandible, and pelvis. Slight bony expansion with endosteal erosion and limited periosteal bone formation may be associated. |
Rare benign neoplasm occurring in the second and third decades of life. Pain and swelling are the leading clinical symptoms, or a pathologic fracture may be the presenting feature. |
Bone island (enostosis)
Fig. 14.35 |
Single or multiple intraosseous foci of homogeneously dense bone most often found in the pelvis, proximal femurs, and spine. They may be round, ovoid, or oblong and are aligned with the long axis of the trabecular architecture. Tiny bone spicules radiating from the periphery of the lesion are characteristic. A somewhat radiolucent center may occasionally be encountered. Range in size from a few millimeters to a few centimeters. |
Incidental finding in all age groups. Lesions may slowly increase or decrease in size over years. Bone scintigraphy is usually negative.
Differential diagnosis: osteoblastic metastases.
A curettaged and methylmethacrylate cemented bone lesion may resemble a large enostosis.
Osteopoikilosis (Fig. 14.36): Numerous small sclerotic foci with symmetric distribution in periarticular locations. |
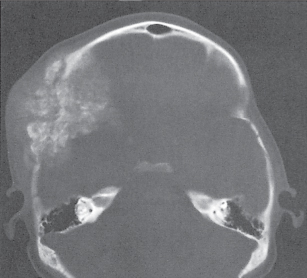
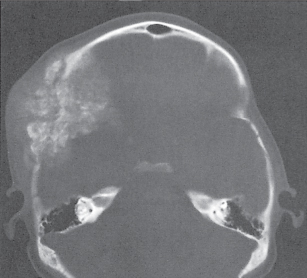
Osteoma
Fig. 14.37a, b
Fig. 14.38 |
Mass of either uniformly dense compact bone or less dense cancellous bone protruding from the skull and facial bones (especially the paranasal sinuses). Rarely, osteomas arise from the cortical surface of the clavicle, pelvis, and tubular bones. |
Gardner syndrome: Autosomal dominant disease consisting of multiple osteomas, colonic polyposis, and soft tissue tumors (especially desmoids).
Tuberous sclerosis: Multiple osteoma-like lesions may be associated, especially in the metacarpals and metatarsals.
Melorheostosis: Partial or circumferential cortical thickening of a tubular bone (”wax flowing down the side of a candle”). Osteoma-like protrusions and soft tissue ossifications may be associated (Fig. 14.39). |
Osteoid osteoma
Fig. 14.40
Fig. 14.41 |
Diaphysis of long tubular bones: Cortical radiolucent lesion (nidus) measuring < 1 cm in diameter surrounded by a zone of uniform bone sclerosis (elliptical thickening of the cortex) is virtually diagnostic. The nidus enhances after intravenous (IV) contrast administration and occasionally has a calcified center.
Intra-articular location: A nonconspicuous small radiolucent focus often without significant reactive sclerosis may be present. A synovial inflammatory response including a joint effusion is commonly associated and may mimic an infectious arthritic process.
Carpal and tarsal bones: A partially or completely calcified lesion with or without only mild reactive sclerosis is characteristic.
Spine: A small osteolytic focus surrounded by extensive sclerosis in the posterior elements is the most common presentation. A scoliotic deformity is usually associated with the lesion that is located near its apex on the concave side of the curvature. |
Occurs in patients between 7 and 25 y of age, with a male predominance (3:1). Pain is the hallmark of the disease, usually more dramatic at night and ameliorated by aspirin. Bone scintigraphy shows an unusual intense uptake in the center of the lesion (nidus) surrounded by the less intense uptake of the adjacent sclerotic bone (double density sign), differentiating it from chronic cortical osteomyelitis with small abscess formation.
Localized cortical thickening caused by a stress fracture typically is associated with a transverse linear radiolucency.
In the lumbar spine, a unilateral osteosclerotic focus about a pedicle is frequently caused by a hyper-trophied interarticular pars caused by unilateral spondylolysis of the contralateral side. |
Osteoblastoma
Fig. 14.42a, b
Fig. 14.43 |
Long tubular bones: Osteoblastomas typically originate in either the medullary or cortical bone of the diaphysis and present as expansile osteolytic lesions, often with areas of calcification or ossification, surrounding bone sclerosis and frequently exuberant solid periosteal reaction.
Spine: Well-defined expansile osteolytic lesion that may be partially or extensively calcified and surrounded by mild sclerosis at best is commonly found in the posterior elements.
Hands, feet, and pelvis: Slightly expansile lytic lesion with varying degrees of matrix calcification/ossification and surrounding reactive sclerosis is the most common presentation. |
Usually diagnosed in the second and third decades of life, with a male predominance (2:1) and similar clinical presentation as in osteoid osteomas.
The size of the nidus (actual size of the osteolytic lesion, which, however, at times may be partially to completely calcified/ossified), is used to differentiate between osteoblastomas (> 2 cm) and osteoid osteomas (< 2 cm).
Aggressive (malignant) osteoblastomas are differentiated from the typical (conventional) osteoblastomas by a more aggressive pattern of tumor behavior, including a much greater likelihood to recur.
The radiologic features of both types are similar, but cortical violation with tumor extension into the neighboring soft tissues is suggestive of an aggressive osteoblastoma. |
Giant cell tumor
Fig. 14.44
Fig. 14.45
Fig. 14.46a–c |
In the long tubular bone (85%), a giant cell tumor presents as an eccentric expansile osteolytic lesion, often with a delicate trabecular pattern (“soap bubble” appearance), extending from the metaphysis to the subchondral bone. The margins of the lesion may be well or poorly defined, but sclerosis and periosteal reactions are typically absent. Cortical breakthrough with spread into the adjacent soft tissues occurs. Fluid levels within the lesion may be evident. Occasionally, the tumor is found in the pelvis, sacrum, ribs, vertebral bodies, hands, and feet. |
Occurs usually in the third and fourth decades of life, with equal gender distribution. Tumor recurrence is about 50% in excised and grafted lesions but markedly reduced when cemented with methylmethacry-late instead of bone grafts.
Malignant giant cell tumors (including malignant transformation of a benign giant cell tumor) account for approximately 5%.
Giant cell (reparative) granuloma has a more benign course and similar histologic and radiologic features as a giant cell tumor, although cortical violation with spread into neighboring soft tissues does not occur, and a predilection for facial bones and short tubular bones of the hands and feet exists. |
Chordoma
Fig. 14.47a, b |
Destructive expansile osteolytic lesion with large soft tissue mass and intratumoral calcifications in up to 90% of cases is characteristic. Calcifications found in the tumor periphery can also represent sequestered bone fragments. Originates from the sacrum and coccyx (60%), clivus (30%), and spine, especially C2 (10%). Sacral lesions typically grow anteriorly and may contain cystic areas. In the spine vertebral collapse, sclerosis (“ivory” vertebrae) and erosion of the posterior aspect of the vertebral body and neural foramina are additional manifestations. Involvement of two contiguous vertebrae is not uncommon. |
Sacrococcygeal chordomas occur in 40- to 60-y-old patients, with male predominance. Spheno-occipital chordomas are usually diagnosed in 20- to 40-y-old patients without gender predilection. The chondroid chordoma is a variant comprising one third of all clivus chordomas with better prognosis and particularly prominent calcifications. Hematogenous metastases may eventually develop in about one third of all chordomas. |
Adamantinoma (angioblastoma) |
Single or multiple, central or eccentric, multilocular, slightly expansile, sharply or poorly delineated osteolytic lesions with or without reactive sclerosis in the diaphysis of the tibia (85%) with preferential involvement of its anterior cortex or, rarely, in other long tubular bones. Cortical destruction, exuberant periostitis, and a soft tissue mass may be associated. |
Usually diagnosed in the third and fourth decades of life with slight male predominance. History of trauma is frequent. Local swelling is the major clinical finding. Differential diagnosis: Osteofibrous dysplasia. See under fibrous dysplasia at the end of this table. |
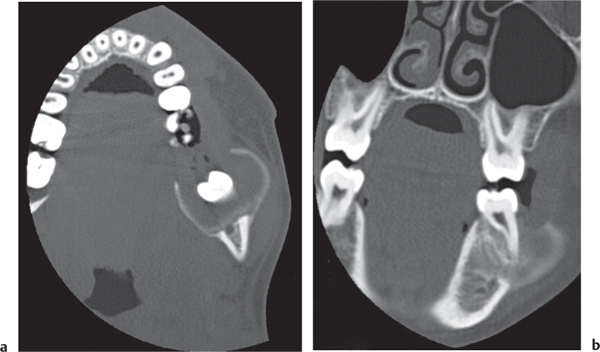
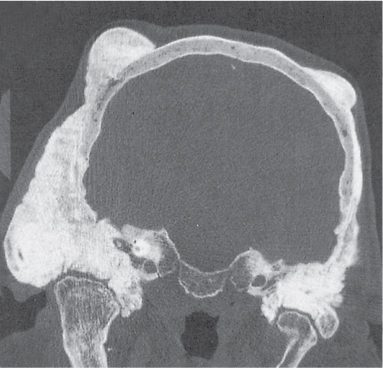
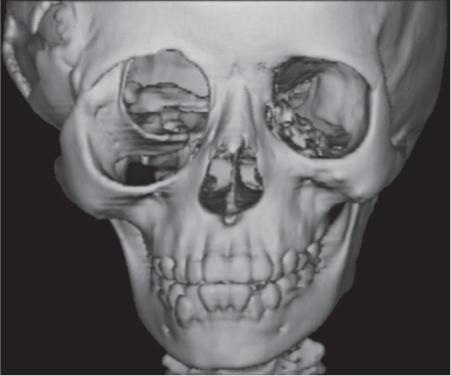
Ameloblastoma
Fig. 14.48a, b |
Presentation ranges from unilocular cyst to multilocular, often trabeculated lesion with cortical expansion or destruction and occasionally a large soft tissue mass in the mandible or, much less commonly, the maxilla. |
Usually diagnosed in the fourth and fifth decades of life without gender predilection. |
Fibrosarcoma
Fig. 14.49 |
Well or poorly marginated osteolytic lesion with general lack of both periosteal reaction and osteo-sclerosis is typical. Irregular thin or thick intratumoral septation, cortical violation, and soft tissue mass are frequently associated. Dystrophic calcifications and sequestered bone fragments are occasionally seen within the tumor. Preferential involvement of the metaphyses of the long tubular bones (70%) with frequent extension into the diaphyses and epiphyses. |
Rare malignant bone tumor occurring in the third to sixth decades of life without gender predilection. The histologic spectrum of fibrosarcomas ranging from well differentiated to poorly differentiated lesions is reflected in the greatly variable radiologic presentation. May complicate Paget disease, bone infarcts, radiation necrosis, and chronic osteomyelitis. |
Malignant fibrous histiocytoma
Fig. 14.50 |
A poorly defined osteolytic lesion with cortical destruction, large soft tissue mass, and absence of both periosteal reaction and new bone formation is the most common presentation. The ends of long tubular bones from diaphysis to epiphysis are affected in 75% of cases with strong predilection for the lower extremity. Osseous expansion is unusual but may be observed in flat bones, such as pelvis, ribs, scapula, and sternum. |
Occurs at any age, but the majority of cases are diagnosed in the fifth, sixth, and seventh decades of life, with slight male predominance. The lesion cannot be radiographically differentiated from a highly malignant fibrosarcoma.
May develop in abnormal bone such as Paget disease and bone infarcts or secondary to radiation therapy. |
Chondrosarcoma, central
Fig. 14.51
Fig. 14.52
Fig. 14.53 |
A slightly expansile, often multilobulated osteolytic lesion with cortical thickening, endosteal erosion, and scattered stippled or irregular calcifications is characteristic. Matrix calcification representing the most specific finding is only present in about two thirds of the cases. Lesions without matrix calcifications are virtually impossible to differentiate from fibrosarcomas. Demonstration of a poorly defined osteolysis, cortical violation, and large soft tissue mass indicate a highly aggressive lesion, but an interrupted periosteal reaction typically is absent. However, more often a geographic destruction pattern is present, suggesting a less malignant process. The tumor has a predilection for the metaphyses of the long tubular bones (especially femur and humerus) and flat bones (especially pelvis) and the vertebrae. |
Occurs usually between 30 and 60 y of age, with male predominance. The tumor arises de novo in the medullary cavity or is caused by malignant transformation of an enchondroma. Low-grade chondrosarcomas are extremely difficult to differentiate from enchondromas. Irregular matrix calcifications (as opposed to punctate, ring, and arclike calcifications), large areas of noncalcified tumor matrix, poorly defined osteolysis, extensive endosteal scalloping, and cortical thickening (even solid) suggest malignancy. |
Chondrosarcoma, peripheral
Fig. 14.54a, b
Fig. 14.55 |
Malignant transformation of a benign osteochondroma is suggested by demonstration of a bulky cartilaginous cap measuring > 2 cm in width, scattered and irregular calcifications within the cartilaginous cap, associated soft tissue mass, focal soft tissue areas within the bony component of the lesion, and destruction or pressure erosion of the adjacent bone. Differentiation of an osteochondroma from a peripheral chondrosarcoma is not always possible with CT, and supplementation with other imaging techniques, such as bone scintigraphy and MRI, is often required. |
Local pain and growth of an osteochondroma in adulthood suggest clinically malignant transformation. The risk of malignant transformation in patients with hereditary multiple exostoses is estimated to be up to 25%, whereas in a solitary osteochondroma, it is < 1%.
Rarely, a peripheral chondrosarcoma develops de novo from the periost (periosteal or juxtacortical chondrosarcoma). |
Clear cell chondrosarcoma
Fig. 14.56 |
Poorly or well-defined (sclerotic borders), slightly expansile epiphyseal lesion with central calcifications in about one third of the cases is characteristic. Most frequent locations are the proximal ends of the femur, humerus, and tibia. |
Rare cartilaginous bone tumor occurring between ages 25 and 50 y, with slight male predominance. Imaging features are virtually identical to a chondroblastoma. |
Dedifferentiated chondrosarcoma |
Occurs in patients older than 50 y without gender predilection. Presents with pain, soft tissue swelling, or pathologic fracture (one third of patients). The prognosis is very poor. |
Features of both a low-grade chondrosarcoma (nonexpansile osteolytic lesion with intact cortex and matrix calcifications) and a highly anaplastic sarcoma (expansile, poorly defined osteolytic lesion with cortical destruction and large soft tissue mass, but usually without calcification) are characteristic. The transition between these two tumor components is usually abrupt. Distribution of the tumor is similar to conventional chondrosarcoma, with femur, humerus, and pelvis being the most frequent sites of involvement. |
Mesenchymal chondrosarcoma
Fig. 14.57 |
Features are indistinguishable from a high-grade conventional chondrosarcoma and include poorly defined osteolysis, matrix calcifications, cortical expansion and violation, and soft tissue mass. Most frequent sites of involvement are femur, ribs, and spine. |
Occurs usually between age 20 and 40 y and has a poor prognosis. Metastases to lymph nodes and other bones are not unusual. Approximately 50% of mesenchymal chondrosarcomas arise in the soft tissues.
Myxoid chondrosarcoma is the most common extraskeletal chondrosarcoma type and has a relatively low-grade malignancy. |
Osteosarcoma
Fig. 14.58a, b
Fig. 14.59
Fig. 14.60
Fig. 14.61 |
Conventional osteosarcoma presenting as a poorly defined intramedullary lesion most often originating in the metaphyses of long tubular bones with varying degrees of osteolysis and osteosclerosis is most characteristic. The tumor may be purely osteolytic or osteoblastic. Cortical destruction associated with an interrupted periosteal reaction in the form of a Codman triangle (laminated triangular elevation of the periost at the tumor periphery) or with perpendicular (“hair on end” or “sunburst”), laminated (“onion skin”), or amorphous appearance is most characteristic. Large soft tissue masses are commonly found in tumors with cortical violation. Surrounding tumor edema and hemorrhage may result in overestimation of the actual tumor size. “Skip” metastases (areas of tumor separated from the main neoplasm) can be identified in the medullary canal by their higher attenuation values than the normal bone marrow. Most common sites of involvement are the long tubular bones (80%), especially the femur, tibia, and humerus. Osteosarcomas are relatively infrequent in the pelvis, spine, and facial bones and rare in the remaining skeleton.
Telangiectatic osteosarcomas commonly present as large, multilocular, expansile, and predominantly lytic lesions, often with cortical violation and tumor extension into the soft tissues, but without significant periosteal reaction and sclerosis. Fluid levels are often also evident.
Small cell osteosarcomas commonly present as large predominantly osteolytic lesions, extending from the metaphysis to the diaphysis of a long tubular bone associated with interrupted periosteal reaction and large soft tissue masses. They resemble Ewing sarcomas.
Intraosseous low-grade osteosarcomas present as purely sclerotic and mixed osteolytic/osteoblastic lesions often limited to the medullary portion of the diametaphyses of long tubular bones, especially of the tibia and femur. Irregular cortical thickening may be associated. The tumor is frequently mistaken for a benign osteosclerotic process. |
Osteosarcomas most commonly present in the second and third decades of life with pain, swelling, restriction of motion, and pyrexia. Men are more frequently affected than women. In the elderly, they may be a complication of Paget disease or radiation therapy.
Gnathic osteosarcomas present as purely osteoblastic, osteolytic, or mixed lesions in the mandible or less frequently maxilla, occur in the middle-aged and elderly, and have a relatively benign course.
Intracortical osteosarcomas are rare and originate in the cortices of the tibia and femur, presenting as slightly expansile osteolytic lesions with surrounding sclerosis.
Osteosarcomatosis refers to simultaneous involvement of more than one skeletal site. It has to be differentiated from a unicentric lesion with distal skeletal metastases. |
Periosteal osteosarcoma |
Oblong, dense lesion arising from the diaphyseal surface of a long tubular bone with irregular thickening and occasionally saucerization of the adjacent cortex is typical. A Codman triangle and radiating or cloud-like osseous proliferation are frequently associated. Femur, tibia, and humerus are the most common sites of involvement. The medullary cavity is typically not involved. |
Usually diagnosed in the second and third decades of life with better prognosis than conventional osteosarcomas, but worse than parosteal osteosarcomas. Radiologic and histologic features of periosteal osteosarcomas and periosteal (juxtacortical) chondrosarcomas are similar if not identical.
A high-grade surface osteosarcoma (Fig. 14.62) arises from the external surface of the cortex with clinical, histologic, and radiologic features comparable to a conventional osteosarcoma. |
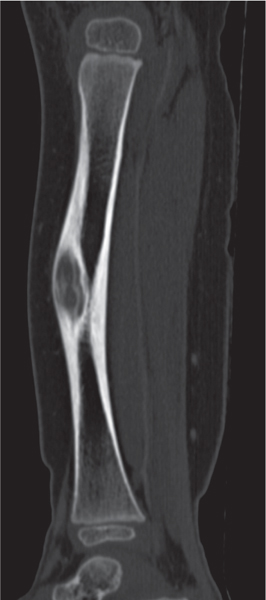
Parosteal osteosarcoma
Fig. 14.63a, b |
A large radiodense oval or round mass with smooth, lobulated, or irregular margins attached in sessile fashion to the external metaphyseal cortex of a long tubular bone is characteristic. A thin radiolucent line may separate the lesion outside its attachment (pedicle) from the cortex, which itself may be thickened. With progressive enlargement, the tumor tends to wrap around the bone. Ossification within the tumor proceeds from its base to the periphery and may be homogeneous or contain radiolucent areas. Tumor extension into the medullary bone is unusual. The most characteristic location is the posterior surface of the distal femur. |
Most common type of osteosarcoma arising on the surface of a bone. Affected patients are adults most often in the third and fourth decades of life. Symptoms are typically insidious and include pain, swelling, and a palpable mass. The prognosis is better than in both conventional and periosteal osteosarcomas.
Differential diagnosis: Posttraumatic myositis ossificans can be differentiated from a parosteal osteosarcoma by the demonstration of a fine radiolucent line separating the lesion in its entire length from the adjacent bone. Furthermore, the ossification in myositis ossificans is evenly radiodense throughout the lesion, and its periphery is sharply delineated, representing cancellous bone surrounded by a cortex. |
Ewing sarcoma
Fig. 14.64
Fig. 14.65 |
A poorly defined osteolytic lesion with cortical violation and large soft tissue mass is characteristic. In tubular bones, an interrupted periosteal reaction of the laminated (“onion skin”) or, less commonly, perpendicular (“sunburst” or “hair on end”) pattern is typically associated. Saucerization of the cortex is a rare but relatively characteristic manifestation of the disease. In flat and irregular bones, osteosclerosis can be the dominant radiographic feature. The tumor can affect any bone but has a predilection for the diametaphysis of long tubular bones and overall the lower half of the skeleton. The most common locations include femur, tibia, humerus, pelvis, and sacrum. |
Usually diagnosed between the ages of 5 and 30 y (peak 10–15 y), with slight male predominance. Involvement of the tubular bones occurs more often in children, whereas involvement of the flat bones is more commonly found in young adults. Patients present with localized pain and swelling, occasionally combined with fever and leukocytosis. The tumor is rare in blacks.
Primitive neuroectodermal tumors (PNETs) (Fig. 14.66a) have similar histologic and imaging features, but epiphyseal involvement, pathologic fractures, and distant metastases occur more frequently when compared with Ewing sarcoma.
Askin tumor is a PNET variant arising from the intercostal nerves or ribs, usually in young Caucasian female patients (Fig. 14.66b). |
Angiosarcoma
Fig. 14.67 |
Solitary or multiple, poorly or well-demarcated lesions most commonly located in the medulla or cortex of long tubular bones, pelvis, spine, and skull. Cortical thinning or violation and/or mild expansion without periostitis is occasionally associated. |
Occurs in the fourth and fifth decades, with male predominance.
If multicentric, it may simulate multiple myeloma, metastases, cystic angiomatosis, and cystic osteomyelitis (e.g., tuberculous or fungal).
Hemangioendothelioma (Fig. 14.68) refers to a low-grade malignant angiosarcoma.
Hemangiopericytoma is a borderline malignant tumor with similar imaging features. |
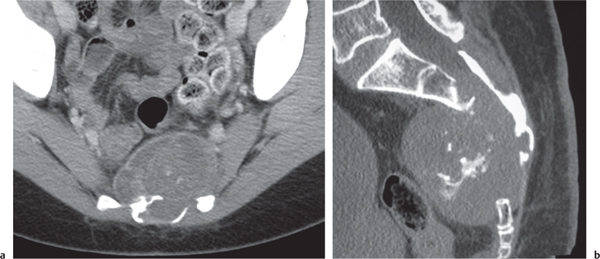
Plasmacytoma
Fig. 14.69 |
Presents either as an expansile (blow-out) trabeculated osteolytic lesion with cortical thinning and violation measuring up to several centimeters in diameter or as a well marginated, purely osteolytic focus with endosteal scalloping. Rarely, it may present as a sclerotic lesion (e.g., “ivory” vertebra). Most common locations are spine and pelvis.
Complications in the spine include transdiscal tumor spread and pathologic fractures resulting sometimes in complete dissolution of a vertebral body. |
Plasmacytoma may be considered a solitary manifestation of multiple myeloma with conversion to the latter occurring as late as 20 y after initial diagnosis. Plasmacytoma affects younger patients than multiple myeloma, with about half the patients being 50 or younger at the time of diagnosis. It frequently presents with neurologic symptoms and radiographically may be mistaken for a giant cell tumor. |
Multiple myeloma
Fig. 14.70a, b |
Multiple well-marginated (“punched out”) or poorly delineated osteolytic lesions of relatively uniform size are characteristic. Endosteal scalloping is typically present with larger lesions in the long tubular bones. Diffuse osteopenia without well-defined areas of osteolysis is, however, the most common presentation. Periosteal new bone formation is exceedingly rare. Focal or multiple sclerotic lesions are an unusual initial presentation but may develop after chemotherapy, irradiation, or pathologic fracture. Preferred locations in order of decreasing frequency are spine, ribs, skull, pelvis, long tubular bones, and clavicles (distal end).
Spine: Generalized osteopenia with or without osteolytic foci is characteristic. Associated findings may include irregular thickening of the vertical trabeculae, relative sparing of the posterior elements, scalloping of the anterior margins of the vertebral bodies (pressure erosions from adjacent soft tissue lesions), and paraspinal or extradural tumor extension. Pathologic compression fractures are a frequent complication. |
Occurs in patients older than 40 y with male predominance, presenting with bone pain, anemia, hypercalcemia, proteinuria (including Bence–Jones proteins), and monoclonal gammopathy (high erythrocyte sedimentation rate [ESR], abnormal electrophoresis). Waldenström macroglobulinemia presents with similar clinical and radiographic findings, but the radiographic features are usually less conspicuous. Plasma cell granuloma: Solitary or scattered, slow-growing osteoblastic foci consisting histologically of a dense infiltrate of normal plasma cells. Considered to be a variant of chronic recurrent multifocal osteomyelitis (CRMO) or SAPHO (synovitis, acne, pustulosis, hyperostosis, and osteitis). POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes): Solitary or multiple osteosclerotic lesions and fluffy spiculated hyperostotic areas preferentially at sites of ligamentous attachments in axial and para-axial locations. |
Metastases
Fig. 14.71a, b
Fig. 14.72
Fig. 14.73
Fig. 14.74 |
Solitary or, more commonly, multiple osteolytic, osteoblastic, or mixed osteolytic-osteoblastic lesions with preferential involvement of the red marrow containing skeleton (spine, pelvis, ribs, skull, proximal femora, and humeri) is characteristic. Osteolytic metastases may be well or poorly marginated. Osteoblastic metastases may be focal or diffuse. In the spine, besides the vertebral bodies, the posterior elements, including the pedicles, are frequently also involved (differential diagnosis: multiple myeloma). Purely osteolytic metastases commonly arise from carcinomas of lung, kidney, thyroid, and lymphoma. Purely osteoblastic metastases are most often associated with prostatic and breast carcinomas, but also with Hodgkin lymphoma, carcinoids, and medulloblastomas. Periosteal reactions are highly unusual in metastatic disease except in prostatic and breast carcinomas, gastrointestinal (GI) malignancies, and neuroblastomas. Neither osseous expansion nor soft tissue masses are typically associated with bone metastases except in rib lesions, osteolytic metastases from carcinomas originating in the kidney, thyroid, lung, and liver, and osteoblastic metastases from prostatic and breast carcinomas. One or more sclerotic vertebral body (“ivory” vertebra) is most often caused by prostatic or breast carcinoma metastases, but also found with Hodgkin lymphoma and Paget disease. Pathologic fractures are a frequent complication, especially in the spine. Response of an osteolytic metastasis to treatment (e.g., chemotherapy or radiation therapy) is evident by progressive sclerosis proceeding from the periphery toward the center of the lesion with eventual reduction in size or disappearance of the osteolytic focus. The appearance of osteosclerotic foci during therapy is usually caused by progression of the disease, but it may also indicate a healing response of preexisting osteolytic metastases that could initially not be identified on imaging examinations. A positive treatment response of osteoblastic lesions is evident by decrease and eventual disappearance of the sclerotic focus. |
Bone metastases are by far the most common skeletal malignancy occurring either by hematogenous spread or direct tumor extension. They most frequently originate from carcinomas of breast, prostate, lung, kidney, thyroid, and GI tract. Breast carcinoma metastases tend to be mixed or, less commonly, purely osteolytic or osteoblastic, often extensive and frequently associated with pathologic fractures. Prostate carcinoma metastases are characteristically osteoblastic, may be expansile (simulating Paget disease), or associated with periostitis. Bronchogenic carcinoma metastases are typically osteolytic or mixed and occasionally expansile. An eccentric erosion of the external cortex of a long tubular bone (“cookie bite” sign) and an intracortical osteolytic lesion are rare in metastatic disease but when present strongly suggest a bronchogenic or GI primary tumor.
Kidney and thyroid carcinoma metastases are often solitary, purely osteolytic lesions and may be expansile and depict a septated (bubbly) appearance. GI tract metastases cover the whole spectrum from purely osteolytic to purely osteoblastic.
Metastases from colon or rectum carcinoma may occasionally resemble an osteosarcoma and depict a sunburst periosteal reaction.
Metastatic bone disease with unknown primary most often originates from prostate, lymphoma, breast, lung, kidney, thyroid, or colon. |
Lymphoma
Fig. 14.75a–d
Fig. 14.76 |
Preferential sites of involvement include spine, pelvis, scapula, and ribs. In long tubular bones, involvement of the diametaphysis of the femur and tibia about the knee is most common.
Non–Hodgkin lymphoma typically presents as solitary or, more often, multiple poorly defined osteolytic lesions.
Histiocytic lymphoma (large B-cell lymphoma) frequently has a mixed osteolytic-osteoblastic appearance and may resemble Paget disease without bony expansion. Purely osteoblastic lesions are uncommon in non–Hodgkin lymphoma.
Hodgkin disease of the bone is caused by secondary involvement. Its presentation ranges from purely osteolytic to purely osteoblastic lesions. Diffuse sclerosis of a vertebral body (“ivory” vertebra) is not an unusual manifestation. Osteolytic lesions tend to be poorly defined and are associated with periostitis in one third of the cases. |
Secondary involvement of bone caused by hematogenous spread or, less frequently, by direct invasion occurs in one third of patients with non–Hodgkin lymphoma and in 10% with Hodgkin disease.
Primary bone lymphoma is much less common, accounting for about 5% of all primary malignant bone tumors, is almost exclusively limited to non–Hodgkin lymphoma (especially the histiocytic [large B-cell] type), and typically occurs in older patients, with a 2:1 male predominance.
Burkitt lymphoma presents as expansile osteolytic lesions associated with a soft tissue mass. Involvement of the facial bones (especially the maxilla) is most characteristic in children in tropical Africa. Nonendemic Burkitt lymphoma may be associated with immunodeficiency (e.g., organ transplantation and acquired immunodeficiency syndrome [AIDS]).
Mycosis fungoides is a T-cell lymphoma with primary involvement of the skin. Discrete or poorly defined osteolytic lesions may be associated in the appendicular skeleton. |
Leukemia |
Diffuse osteopenia with medullary widening and cortical thinning in tubular bones and vertebral compressions are the most common presentation. Moth-eaten or permeative osteolysis may be found in both tubular and flat bones. Radiolucent and/or radiodense metaphyseal bands, as well as periosteal new bone formation, are particularly common in children. Complications may include intra-articular and subperiosteal hemorrhages, septic arthritis, osteomyelitis, osteonecrosis, and secondary gout.
Granulocytic sarcomas (chloromas) present as single or multiple, often expansile lytic lesions in the skull, spine, ribs, sternum, and long tubular bones, usually in acute myelogenic leukemia. |
Leukemias are classified based on cell maturity (acute with immature or blastic cells versus chronic with mature cells), cell morphology (myeloid versus lymphoid) or cell origin (thymus derived T cells versus blood marrow derived B cells).
Skeletal abnormalities are similar for all forms of leukemias, but are most common in acute childhood leukemias, where they occur in over 50% of cases, and least common in chronic leukemias. |
Langerhans cell histiocytosis (histiocytosis X)
Fig. 14.77
Fig. 14.78
Fig. 14.79a–c |
Eosinophilic granuloma presents as solitary or, less commonly, multiple lesions with preferential involvement of skull, mandible, spine, ribs, and the diametaphyses (rarely epiphyses) of long tubular bones. Relatively well-defined radiolucent areas with endosteal scalloping with or without slight bone expansion and varying degrees of periosteal new bone formation and sclerosis are typical in the long bones. More aggressive lesions with cortical violation and interrupted laminated periosteal reaction may mimic acute osteomyelitis or Ewing sarcoma. Well-defined lytic lesions with or without sclerotic borders may be found in the skull and pelvis. A radiodense focus (button sequestrum) is frequently observed in skull lesions. Larger osteolytic areas in the skull typically depict beveled edges caused by the uneven destruction of the inner and outer tables. In the mandible, radiolucent lesions about the teeth may lead to the “floating teeth” appearance. In the spine, a collapsed vertebral body (vertebra plana) with intact intervertebral spaces or, less frequently, a lytic and occasionally slightly expansile lesion involving the vertebral body and/or the posterior elements may be found. |
Langerhans cell histiocytosis comprises three major conditions.
1. Eosinophilic granuloma is both the most common and most benign variant, representing about 70% of cases. It is usually diagnosed between age 5 and 20 y.
Spontaneous healing of a solitary lesion occurs, typically progressing from the periphery toward its center, and eventually resulting in its disappearance or transformation into a sclerotic focus.
2. Hand–Schüller–Christian disease is characterized by the triad of exophthalmos, diabetes insipidus, and large lytic skull lesions (“geographic skull”).
3. Letterer–Siwe disease is the acute disseminated variant in children younger than 2. Bone lesions are less common but may include multiple widespread lytic lesions in the skull (“raindrop” pattern). Hepatosplenomegaly, lymphadenopathy, and nonitching eczematous skin lesions are commonly associated. |
Amyloidosis |
Osteolytic lesions of variable size with endosteal scalloping simulating multiple myeloma preferentially located in the proximal humerus or proximal femur is the most common presentation. Subchondral amyloid deposition may result in avascular necrosis caused by perivascular amyloid deposition with subsequent vascular occlusion. Pathologic fractures are a relatively common complication. Subchondral cyst formation and erosions in the hand and wrist (especially carpal bones) associated with periarticular or diffuse osteoporosis may simulate rheumatoid arthritis, although extensive nodular soft tissue masses, well-defined cystic lesions with or without surrounding sclerosis and preservation of the joint space are more characteristic for amyloidosis. |
Musculoskeletal abnormalities are the result of amyloid deposition in bone, synovium, and soft tissue. Primary amyloidosis occurs in patients older than 40, with male predominance. It may be associated with multiple myeloma.
Secondary amyloidosis is associated with chronic renal disease, rheumatoid arthritis, lupus erythematosus, ulcerative colitis, chronic suppurative disease, and lymphoproliferative disorders. |
Brown tumor in hyperparathyroidism
Fig. 14.80 |
Single or multiple, occasionally expansile, well- to poorly defined osteolytic lesions of the axial and appendicular skeleton. Eccentric or cortical location is not unusual. Common sites of involvement are facial bones, pelvis, ribs, and femora. Brown tumors may undergo necrosis and liquefaction, producing cysts, or with proper treatment (removal of the parathyroid adenoma) become increasingly sclerotic. |
Brown tumors are more commonly associated with primary than secondary hyperparathyroidism. Other manifestations of hyperparathyroidism are usually also apparent and include osteopenia, subperiosteal, endosteal, and subchondral bone resorption, intracortical tunneling, chondrocalcinosis, and paraarticular calcifications and vascular calcifications. Bone sclerosis is common in secondary hyperparathyroidism. |
Hemophilic pseudotumor
Fig. 14.81 |
Central (intraosseous) or eccentric (subperiosteal), well-demarcated osteolytic lesion, often associated with cortical violation, a solid or interrupted periosteal reaction, and a large soft tissue mass is a common presentation. Minimal to massive calcification within the lesion is occasionally also encountered. Rarely, more than one bone contains a pseudotumor. Preferred locations are pelvis, femur, tibia, and hand. |
Lesions are late sequelae of intramedullary or periosteal hemorrhage/hematoma occurring in < 2% of hemophiliacs.
Hemophilic arthropathy, including dense joint effusions and joint contractures, avascular necrosis, especially of the femoral head and talus, spontaneous fractures, and soft tissue hematomas, may also be evident. Knee, ankle, and elbow are most frequently affected. |
Intraosseous tophus in gout |
One or more cystic lesions often with partial calcification may be found in the subchondral and deeper osseous areas simulating enchondromas. Larger intraosseous calcifications may mimic bone infarcts. These findings are most frequently seen in the hands and feet. Association with characteristic findings of gouty arthropathy is diagnostic. |
Occurs in about 5% of patients with chronic gouty arthritis. Intraosseous urate deposition with subsequent calcifications usually originates from the adjacent joint, penetrates the cartilaginous surface, and extends into the spongiosa. |
Osteonecrosis (bone infarct and avascular necrosis)
Fig. 14.82 |
Early signs are nonspecific and include mottled osteopenia, poorly defined osteolytic lesion (s), or patchy osteopenic and sclerotic areas.
Findings are more characteristic in a more advanced later stage. In the diametaphyses, they include serpiginous peripheral rim of calcification or sclerosis surrounding an oblong area of bone rarefaction. Periostitis and matrix calcifications are frequently associated. Intramedullary calcifications are often the only finding and are typically shell-like and peripheral, whereas the calcifications in chondroid matrix tumors tend to be punctate, ringlike or irregular and central and are surrounded by a rim of noncalcified, radiolucent tumor matrix. Solid periosteal reactions and cortical thickening may be associated with both conditions, but in the case of chondroid matrix tumor suggest a low-grade chondrosarcoma rather than enchondroma. Typical findings of osteonecrosis in the epiphyses (avascular necrosis) include curvilinear subchondral sclerosis, subchondral cyst (s) with sclerotic rim, arclike subchondral radiolucency (crescent sign), subchondral fragmentation, and eventually collapse of the articular surface, with considerable sclerosis and secondary degenerative changes in the affected joint. |
Osteonecrosis can be divided into bone infarction, occurring more frequently in the metadiaphyseal regions of long bones (e.g., femur, humerus, and tibia) than in the axial skeleton, and avascular necrosis involving the subarticular bone. Solitary bone infarcts are frequently diagnosed as an incidental finding. Scintigraphy and MRI are far more sensitive than CT in the diagnosis of early avascular necrosis. Osteonecrosis may be idiopathic (25%) or associated with hematological and reticuloendothelial diseases (e.g., sickle cell and Gaucher disease), connective tissue diseases (e.g., lupus erythematosus), trauma (e.g., subcapital hip fracture), human immunodeficiency virus [HIV] infection, renal transplantation, alcoholism, steroid use, pancreatitis, gout, amyloidosis, hemophilia, irradiation, and caisson disease. Radiation osteitis (osteonecrosis) (Fig. 14.83) presents with a mixture of osteopenia, osteosclerosis, and coarse trabeculation (pagetoid bone). Round to ovoid radiolucent lesions within the cortex of long tubular bones are characteristic as opposed to endosteal scalloping caused by tumor recurrence. Bony changes are dose-dependent, with a minimal dose of 3000 cGy required, and occur at the earliest 1 y after irradiation, at a time, when a nuclear medicine scan no longer depicts an increased uptake. Calcified intraosseous hematomas may present on CT similar to a calcified bone infarct. |
Healing fracture/stress fracture
Fig. 14.84 |
Osteosclerosis and localized callus formation in the cortex of long tubular bones may simulate chronic cortical osteomyelitis and osteoid osteoma, respectively. Demonstration of a fracture line in an area of localized elliptical cortical thickening of a long tubular bone (as opposed to a round or ovoid intracortical abscess or a nidus) is diagnostic. |
CT is useful in the assessment of fracture healing and the diagnosis of complications, such as delayed union, nonunion, osteomyelitis, osteonecrosis, and posttraumatic myositis ossificans. |
Pigmented villonodular synovitis (PVNS)
Fig. 14.85 |
Intra-articular/periarticular soft tissue mass and joint effusion with often slightly increased attenuation values due to varying degrees of hemosiderin deposition are characteristic. Subchondral pressure erosions with or without sclerotic margins may be found in “tight” joints, such as hip, ankle, and wrist. Calcifications, osteoporosis, and joint space narrowing are typically absent. Pressure erosions in the knee (most commonly involved joint) are uncommon. |
Monoarticular proliferative synovial disorder presenting with insidious onset of swelling, pain of long duration, and decreased range of motion occurring typically in young adults. A hemorrhagic (chocolate brown) joint effusion in the absence of trauma is characteristic.
Differential diagnosis: synovial (osteo) chondromatosis in which calcified/ossified loose bodies are characteristic. |
Osteomyelitis (acute)
Fig. 14.86
Fig. 14.87
Fig. 14.88
Fig. 14.89 |
Hematogenous osteomyelitis presents initially in the medullary space with focal osteoporosis, endosteal scalloping, and osteolysis due to hyperemia, edema, abscess formation, and trabecular destruction. Progression of the disease to the Haversian and Volkmann canals results in cortical fissuring with subsequent destruction and subperiosteal abscess formation. A periosteal reaction (laminated or, less commonly, spiculated) is a characteristic finding at this stage. After 1 month, a sequestrum (detached necrotic cortical bone) presenting as a radiodense bony spicule surrounded by granulation tissue and newly formed cortical bone (involucrum) may be evident. An opening in the involucrum is termed a cloaca. Sinus tracts leading to the skin surface are often evident. Occasionally, intraosseous gas or fat–fluid (pus) levels may be seen. In both infants and adults, extension of the disease process into the adjacent joint is common, whereas in childhood (1–16 y), the open growth plate represents an effective barrier to the spread of the infection from the metaphysis to the epiphysis and joint, respectively.
Osteomyelitis from a contiguous soft tissue infection typically presents as focal osteoporosis due to edema with subsequent cortical erosion. Periostitis is another less frequent initial finding. Cortical destruction and bone marrow infection with abscess formation may ensue. |
Affects all ages, but is commonly found in children, diabetics, and IV drug abusers. Occurs by hematogenous route, spread from contiguous infection, or direct implantation (punctures, penetrating injury, and postoperative infection). Pyogenic osteomyelitis in children is most often caused by Staphylococcus aureus, Streptococci, Escherichia coli, and Haemophilus influenzae. Gram-negative organisms are not uncommon in adults and IV drug abusers.
Tuberculous osteomyelitis occurs mainly by the hematogenous route. Spinal involvement accounts for about 50% of skeletal tuberculosis. Paraspinal abscess formation (e.g., in psoas muscle) is common and frequently contains calcifications. Compared with pyogenic osteomyelitis, osteoporosis is more pronounced, whereas new bone formation is less extensive.
Tuberculous dactylitis (spina ventosa) refers to cystic expansion of the short tubular bones of the hands and feet of young children with varying degrees of periostitis.
Cystic tuberculosis presents as one or multiple well-defined osteolytic foci without sclerosis, preferentially in the peripheral skeleton.
Fungal osteomyelitis resembles a tuberculous infection. Solitary or multiple osteolytic lesions with discrete margins, mild surrounding sclerosis, and little or no periosteal reaction are a common presentation. |
Brodie abscess
Fig. 14.90 |
Usually solitary, lytic, and often elongated lesion with sclerotic border typically in the metaphyses of long bones. Epiphyses, diaphyses, and flat and irregular bones (e.g., carpus and tarsus) are less common locations. In the epiphysis, a circular well-defined osteolytic lesion is typical. In the diaphysis, the abscesses may be found in central, subcortical, or cortical locations. In the cortex, the abscess is surrounded by periosteal new bone formation, simulating an osteoid osteoma or a stress fracture. |
Subacute pyogenic osteomyelitis (smoldering indolent infection), usually of staphylococcal origin, is common in children, in whom the lesion is typically located in the proximal or distal tibia metaphysis and sometimes connected to the growth plate by a tortuous channel. Histologically, a central purulent or mucoid fluid collection is surrounded by inflammatory granulation tissue and spongy bone eburnation. The lesion may occasionally contain a central sequestrum. |
Osteomyelitis, chronic
Fig. 14.91 |
Thick, irregular sclerotic bone with radiolucencies and extensive periosteal new bone formation is characteristic. Signs of remaining activity or reactivation include a change from the previous exam, poorly defined areas of osteolysis, laminated periosteal reaction, poorly defined bony excrescences, and demonstration of a sequestrum, sinus tract, or soft tissue abscess.
Sclerosing osteomyelitis of Garré is a low-grade infection without purulent exudate presenting as focal or circumferential cortical thickening and sclerosis in the mandible (most commonly) or diaphyses of long tubular bones. In the latter location, osteoid osteoma, stress fracture, and Ribbing disease (hereditary multiple diaphyseal sclerosis with typically asymmetric distribution) must be considered in the differential diagnosis. |
Late acquired syphilis resembles chronic osteomyelitis. Thickened sclerotic long bones caused by endosteal and periosteal new bone formation and ill-defined lytic lesions (gumma formation) are characteristic.
CRMO (chronic recurrent multifocal osteomyelitis, chronic plasma cell osteomyelitis) presents most commonly in children with a protracted clinical course and often symmetric involvement of the medial ends of the clavicles or the metaphyses of long bones with a combination of osteolysis and osteosclerosis and extensive periosteal reaction and new bone formation.
SAPHO syndrome may be a related condition.
Epidermoid carcinoma occurs in 1% of osteomyelitis at the site of a chronically draining sinus and is evident as an enlarging soft tissue mass eroding the osteomyelitic bone. |
Gorham disease (vanishing bone disease)
Fig. 14.92 |
Progressive, often massive osteolysis without attempt of repair (no periosteal or osteosclerotic reaction) spreading across joints or intervertebral spaces is characteristic. Preferential locations are the hip and shoulder regions, although any bone can be affected. Tapering or “pointing” of the long bones at the sites of osteolysis is typical.
In the pelvis, a rapidly destructive arthropathy of the hip presenting with destruction of primarily the femoral head and to a lesser degree the acetabulum must be differentiated. This condition may represent an unusual aggressive form of osteoarthritis (rapidly progressive destructive osteoarthritis), osteonecrosis, CPPD, or hydroxyapatite crystal deposition disease (HADD), or may occur after intra-articular corticosteroid injections.
Proximal femoral focal deficiency (PFFD) characterized by partial absence of the proximal femur is diagnosed in infancy. |
Occurs sporadically at any age, without gender predilection, but is usually diagnosed before the age of 40 y. Histologically, a nonmalignant proliferation of angiomatous and fibrous tissue is evident.
Other osteolysis syndromes diagnosed in infancy or childhood include acro-osteolysis of Hajdu and Cheney, Joseph, or Shinz, carpal-tarsal osteolysis (hereditary or associated with nephropathy), Farber disease (elbows, wrists, knees, and ankles), and Winchester syndrome (carpal and tarsal areas and elbows). |
Fibromatosis |
Solitary or multiple soft tissue masses with erosions of the adjacent bone or osteolytic lesions. Cortical hyperostosis may be associated with bone involvement. |
Variety of benign, but often aggressive fibrous proliferations presenting in both children and adults as tumorlike soft tissue lesions. Involvement of the adjacent bone is not uncommon in infantile forms of the disease. |
Membranous lipodystrophy (polycystic lipomembranous osteodysplasia) |
Symmetric and slowly progressive lytic lesions are found in the carpal and tarsal bones and the ends of long and short tubular bones. Bone deformities may eventually occur from pathologic fractures. |
Rare hereditary disorder of adipose tissue affecting primarily the bones and brain. Presents commonly in the second or third decade of life with painful bones and joints and subsequently with presenile dementia. |
Paget disease (osteitis deformans)
Fig. 14.93
Fig. 14.94
Fig. 14.95
Fig. 14.96 |
Monostotic or more commonly polyostotic asymmetric lesions with preferential involvement of the pelvis, femur, tibia, spine, skull, scapula, and humerus. The pattern of involvement ranges from purely osteolytic (e.g., osteoporosis circumscripta in the calvarium, and V- or flame-shaped defect in the diaphyses of long bones) to purely osteosclerotic (e.g., “ivory” vertebra). The mixed pattern (e.g., “picture frame” vertebra and “cotton wool” appearance of the skull) that is usually associated with bony enlargement, cortical thickening, and coarse trabeculation represents the most common manifestation. In long bones, the disease characteristically progresses from an epiphysis to the diaphysis. Bone softening may result in bowing deformities of the long bones, acetabular protrusion, biconcave compression of vertebral end plates, and basilar invagination. Complications include cortical stress fractures, single or multiple horizontal radiolucent lines in the convex aspect of the deformed bone (lateral aspect of the femoral neck and shaft, anterior aspect of tibia), transverse pathologic fractures, and sarcomatous degeneration. |
Common disorder of middle-aged and elderly patients that is often diagnosed as an incidental finding on radiographs obtained for unrelated purposes. The disease is present in 10% of patients over the age of 80 y and rare in patients under 40.
Laboratory findings include elevated alkaline phosphatase and hydroxyproline levels in the serum and abnormally high hydroxyproline urine levels. Serum acid phosphatase values are normal.
Neoplastic involvement in Paget disease includes sarcomatous degeneration, giant cell tumor, aneurysmal bone cyst, and superimposition of metastases, multiple myeloma, and lymphoma. Sarcomatous degeneration occurs in about 1% of patients usually older than 55 y. Predominantly osteolytic osteosarcomas (60%), malignant fibrous histiocytoma/fibrosarcomas (25%), and chondrosarcomas (10%) are the most frequent tumors found in sarcomatous degeneration.
Calvarial hyperostosis interna (frontalis or generalisata) must be differentiated from Paget disease. Both conditions affect primarily the inner table, but in calvarial hyperostosis, it is irregularly thickened or nodular in outline and predominates in elderly women (Fig. 14.97). |
Fibrous dysplasia
Fig. 14.98
Fig. 14.99a, b
Fig. 14.100
Fig. 14.101
Fig. 14.102 |
Solitary or multiple, often slightly expansile radiolucent lesions that may have a hazy quality (“ground glass” appearance). The matrix may also be uniformly dense, partially calcified or ossified, or thick, dense bands may be present. A curvilinear sclerotic rim may outline lytic lesions. Purely sclerotic lesions occur in the skull and facial bones. The monostotic form (75%) commonly involves a rib, femur, tibia, humerus, and mandible, whereas the polyostotic form (25%) frequently involves the skull and facial bones, pelvis, spine, and shoulder girdle besides the long tubular bones. Polyostotic fibrous dysplasia may be unilateral or bilateral and may affect several bones of a single limb, both limbs, or all four extremities besides the axial skeleton. Solitary lesions in the long bones are located in the diaphyses or, less commonly, metaphyses. Bowing deformities are frequent in the polyostotic form. |
Usually diagnosed before the age of 30.
In McCune-Albright syndrome, precocious female sexual development and café-au-lait spots are associated with the polyostotic form. Differential diagnosis: Neurofibromatosis, where more irregularly contoured and darker café-au-lait spots are frequently associated with similar bone lesions.
Ossifying fibromas are closely related to fibrous dysplasia and occur in the facial bones (especially mandible) and tubular bones (especially anterior aspect of the tibia). In the latter location, they are also referred to as osteofibrous dysplasia (Figs. 14.103 , 14.104 [ p. 538 ] ).
Skeletal abnormalities in neurofibromatosis 1 (von Recklinghausen disease) may simulate polyostotic fibrous dysplasia. A unilateral enlarged orbit with absent lesser and greater sphenoid wings is diagnostic (Fig. 14.105 , p. 538). Severe kyphoscoliosis, associated with vertebral body wedging and scalloping, and foraminal enlargement are typical findings in the spine. Congenital pseudoarthrosis in the distal tibia and fibula is another manifestation. |
Sarcoidosis |
Solitary or multiple osteolytic lesions with a coarsened or lacework trabecular pattern that may be associated with endosteal scalloping or cortical violation without periosteal reaction is the most common presentation. Purely osteolytic (“punched out”) cystic lesions or purely osteosclerotic foci are less common presentations. The hand is the predominant site of involvement where acrosclerosis or acro-osteolysis may also be present. |
Osseous sarcoidosis is usually associated with either skin lesions or pulmonary disease. |
Particle disease (foreign body granuloma)
Fig. 14.106 , p. 538 |
Presents typically as scalloped osteolytic lesions with or without sclerotic margins about a prosthetic component. Larger expansile lesions may become trabeculated and eventually break through cortex without inciting a periosteal reaction. |
Foreign body reaction to prosthetic components such as polymethylmethacrylate cement, polyethylene, and silicone. Rarely, an intraosseous foreign body granuloma is found that is not associated with joint replacement surgery or any other known cause of accidental foreign body implantation. If the foreign body is located in the cortex, the lesion may mimic a chronic cortical abscess or osteoid osteoma/osteoblastoma, respectively. |