Diagnosis: Sporadic Creutzfeldt–Jakob disease (sCJD)
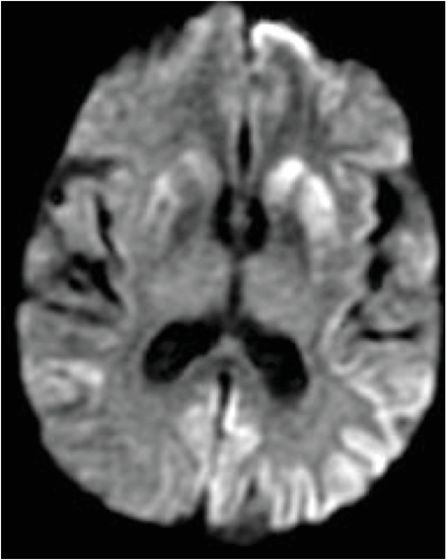
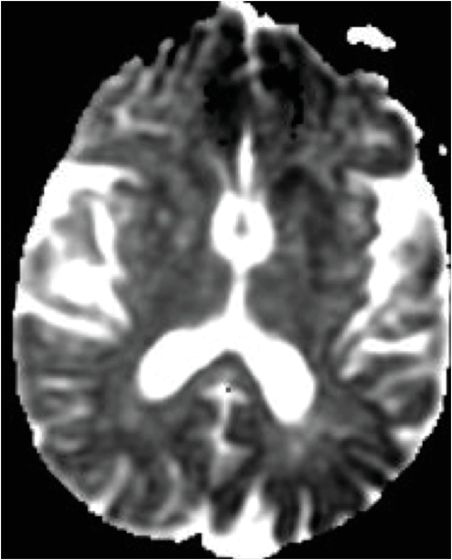
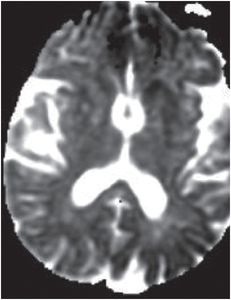
Axial FLAIR (left image) reveals mild putaminal and caudate hyperintensity (yellow arrows), and subtle hyperintensity of the left parietal cortical ribbon (red arrow). Axial DWI (middle image) reveals hyperintensity of the putamina and caudate heads (yellow arrows), and patchy areas of cortical hyperintensity most pronounced in the left parietal lobe (red arrows). ADC map (right image) reveals corresponding hypointensity indicating that signal abnormality on the diffusion-weighted image represents reduced diffusion rather than T2 “shine through.”
Discussion
Overview of Creutzfeldt–Jakob disease
Creutzfeldt–Jakob disease (CJD) is a prion disease that is classified as a transmissible spongiform encephalopathy. It may be sporadic, familial, or iatrogenically spread via contaminated surgical instruments, human growth hormone therapy, cadaveric dural grafts, and corneal implants.
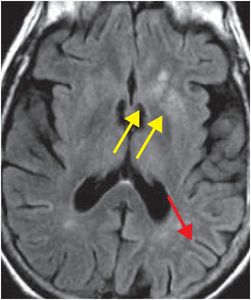
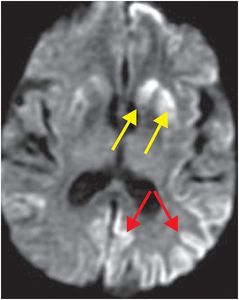
The term “variant CJD” refers to bovine spongiform encephalopathy (“mad cow disease”). This entity characteristically involves the pulvinar nuclei of the posterior thalami, and thus, has a different appearance from “non-variant” etiologies. Signal abnormality in the pulvinar and dorsomedial thalami seen in variant CJD characteristically produces the “hockey stick” sign on FLAIR imaging.
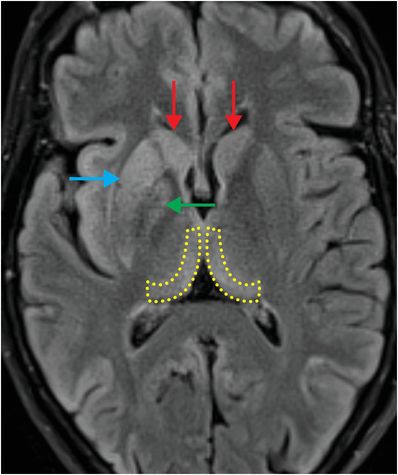
Axial FLAIR MR in a patient with variant CJD demonstrates symmetrical dorsomedial thalamic and pulvinar hyperintense signal bilaterally, producing the “hockey-stick” sign (yellow dashed outlines). There is also hyperintense signal in the cortex, bilateral caudate (red arrows), right putamen (blue arrow), and right globus pallidus (green arrow).
Clinical presentation of CJD
Patients are most commonly between 50 and 75 years of age, and typically present with myoclonus, akinetic mutism, and dementia that is rapidly progressive over several months.
Occasionally, clinical presentation is atypical, with dementia that has a protracted but progressive course, worsening over several years.
Later in the disease course, EEG may show “periodic sharp wave” complexes.
Cerebrospinal fluid analysis may reveal the presence of 14–3–3 protein, but this finding is neither sensitive nor specific. Some cases are diagnosed by brain biopsy or autopsy.
Virtually all cases are fatal within 1 year of diagnosis.
Imaging findings of CJD
CJD is distinguished from other neurodegenerative diseases by the characteristic appearance of restricted diffusion in the cortex and basal ganglia on diffusion-weighted MRI.
Involvement of the brain may be patchy and asymmetric.
Imaging differential diagnosis of CJD
On imaging, CJD may appear similar to various conditions, such as arterial or venous infarction, toxic encephalopathy (e.g., from methanol poisoning), mitochondrial disease, paraneoplastic syndrome, and viral encephalitis.
Specific clinical and imaging findings may be used to differentiate among these entities.
Clinical synopsis
This 74-year-old woman with sporadic CJD and rapidly progressive dementia died several months after diagnosis.
Self-assessment
|
|
|
|
|
|
Related posts:
 12 68-year-old man with left lower quadrant pain and hypotension
12 68-year-old man with left lower quadrant pain and hypotension
 72 67-year-old female with shoulder pain and limited range of motion following a fall onto an outstretched hand
72 67-year-old female with shoulder pain and limited range of motion following a fall onto an outstretched hand
 35 37-year-old woman with a history of rheumatoid arthritis presenting with non-resolving bilateral effusions and chest pain
35 37-year-old woman with a history of rheumatoid arthritis presenting with non-resolving bilateral effusions and chest pain
 62 38-year-old male complaining of diffuse abdominal pain after a motor vehicle collision
62 38-year-old male complaining of diffuse abdominal pain after a motor vehicle collision
 51 90-year-old male with elevated blood pressure and change in mental status. No history of trauma
51 90-year-old male with elevated blood pressure and change in mental status. No history of trauma
 66 21-year-old male with quadriplegia after diving into a shallow pond. The patient struck his head against an embankment, with his head flexed, chin against chest
66 21-year-old male with quadriplegia after diving into a shallow pond. The patient struck his head against an embankment, with his head flexed, chin against chest
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


