6 Orbit and Globe(Table 6.3 – Table 6.4)
Disease | CT Findings | Comments |
Optic nerve–sheath complex lesions | See Table 6.2 |
|
Inflammatory/infectious conditions | ||
Idiopathic orbital inflammatory disease (orbital pseudotumor) | CT is typically nonspecific and variable because the disease process, which is usually unilateral (75%) and multifocal, may present in many forms: • Enlargement of the extraocular muscles (myositic type) with involvement of any part of one or multiple extraocular muscles, including the tendinous insertion anteriorly, and with inward bowing of the medial contour of the muscle belly. Spillover of the inflammatory process into the orbital fat bordering the muscle makes the margin of the muscle indistinct or irregular rather than sharply marginated. • Diffuse oblong enlargement of lacrimal gland with moderate enhancement • Retrobulbar mass lesion (tumefactive type) with soft tissue density, irregular margins, and marked contrast enhancement, which may extend across multiple compartments, sometimes with extraorbital extension, but without bone destruction • Diffuse ill-defined infiltrations with obliteration of retrobulbar fat planes • Diffuse enlargement of the optic nerve–sheath complex (38%) with enhancement of the sheath contrasting against the central low density of the nerve • Uveal-scleral thickening (periscleritis) usually with extension along the most anterior portion of the optic nerve • Apical involvement with abnormal enhancement at the orbital apex extending into the enlarged cavernous sinus • Sclerosing pseudotumor with diffuse increase in density of the orbital fat with obliteration of the optic nerve, muscles, and circumferential involvement of the globe with complete fixation of the intraorbital structures. | Pseudotumor is the most common painful orbital mass in adults and the third most common ophthalmic disorder (5% of all orbital lesions). Orbital pseudotumor can involve retrobulbar fat (76%), extraocular muscles (57%), optic nerve–sheath complex (38%), uveal-scleral area (33%), and lacrimal gland (5%). Wegener granulomatosis, polyarteritis nodosa, lupus erythematosus, rheumatoid arthritis, sarcoidosis, fibrosing mediastinitis, retroperitoneal fibrosis, sclerosing cholangitis, primary orbital vasculitis, thyroiditis, or lymphoma can mimic this nongranulomatous acute, subacute, or chronic inflammatory process in the orbit. Presents with rapidly developing, painful ophthalmoplegia, proptosis, and chemosis. Optic nerve dysfunction results from inflammation of the perineural tissue or compression on the optic nerve from mass effect. Rapid and lasting response to steroid therapy is characteristic. No gender preference. Peak incidence: fifth decade. Pediatric pseudotumor encompasses 6% to 16% of orbital pseudotumors. A regional variant of pseudotumor is the Tolosa–Hunt syndrome, which is characterized by inflammatory infiltration on the orbital apex, including superior orbital fissure and cavernous sinus. It presents with a painful ophthalmoplegia (CN III, IV, and VI) and hypoesthesia of the periorbital skin. |
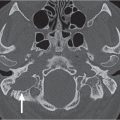
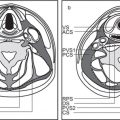
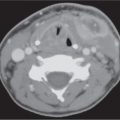
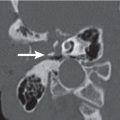
Orbital cellulitis and abscess formation | Predisposing conditions such as sinusitis, foreign bodies, or fracture are usually evident. In preseptal inflammation, CT reveals a diffuse increase in density and thickening of the lid and conjunctiva. Low attenuation areas with or without rim enhancement reflect abscesses in the lid. In subperiosteal accumulation of inflammatory infiltrate or pus, usually originating from the ethmoid sinus, the medial rectus muscle is displaced laterally, often enhances slightly, and is thickened from the inflammatory edema. An enhancing rim indicates the displaced periosteum. A low-density area either localized or extending along the entire medial orbital wall reflects pus accumulation in the subperiosteal space. Demineralization, thinning, or loss of bone manifests inflammation or localized osteomyelitis of the lamina papyracea. Predisposing conditions such as sinusitis, foreign bodies, and fractures are usually evident. Extension of the infection into the intraconal space is manifested on CT by an ill-defined infiltrate with increased attenuation of the orbital fat (stranding and streaking giving rise to “dirty” appearance of the intraconal fat), and obliteration of the enhancing and swollen extraocular muscles and optic nerve–sheath complex. A low-density area with rim enhancement reflects an abscess. Air may be visible within the infiltrate caused either by anaerobic bacteria or leakage of air from the sinuses into the orbit. In diffuse extraconal and intraconal orbital infection, the least common type of orbital involvement, the entire orbital cavity, including the muscles and optic nerve, is obliterated by a diffuse increase in density with concomitant severe proptosis. In cases of severe inflammatory disease of the orbit, periorbital extension to the facial structures and the temporal and infratemporal fossae may occur. Posterior orbital extension may lead to superior orbital fissure syndrome, cavernous sinus thrombosis, and other intracranial complications. | Orbital cellulitis is a serious pyogenic infection of the orbit. It is most frequently the result of bacterial spread from contiguous paranasal sinus (particularly ethmoid) infection (84%). Other sources of infection are primary infections of the face, teeth, or pharynx, septicemia, retrograde thrombophlebitis, trauma, and penetrating foreign bodies. The patient is acutely ill with fever, ocular pain, lid edema, chemosis, proptosis, and limited ocular movements. Fungal infections (mucormycosis, aspergillosis) may occur as a complication of diabetic ketoacidosis and in immuncompromised and debilitated patients. They demonstrate a much more aggressive course with early bone destruction and invasion of arteries and veins. |
Sarcoidosis | Ophthalmic manifestations of sarcoidosis may consist of • Unilateral or bilateral isodense enlargement or mass-like infiltration of the lacrimal glands and extraocular muscles, with contrast enhancement • Optic nerve–sheath complex thickening with abnormal enhancement of the optic nerve from the globe to the chiasm • Pseudotumor-like intraorbital enhancing masses Intracranial extension leads to leptomeningeal, dural, and parenchymal neurosarcoidosis with pathologic enhancement. | Systemic disease of unknown etiology characterized by noncaseating, granulomatous inflammation. It has a particular proclivity for adults younger than age 40 (slight female predominance) and for certain ethnic and racial groups (increased in African descent, Puerto Rican, Irish, and Scandinavian). Ophthalmic manifestations develop in ~25% of patients. In addition to the globe (uveitis, retinal vasculitis, and vitritis), the conjunctiva, extraocular muscles, retrobulbar space, lacrimal gland, optic nerve, chiasm, and optic radiation may be affected. The clinical signs consist of pain, proptosis, ophthalmoparesis, and visual loss. |
Wegener granulomatosis | Characteristically, ocular and orbital involvement is bilateral. The CT appearance is similar to orbital pseudotumor with diffuse retrobulbar infiltration but with bony destruction. Wegener granulomatosis can affect the anterior optic pathways with infiltration of the optic chiasm and with enhancing lesions in the chiasmatic cistern, enhancement of the basal meninges, and thickening of the pituitary infundibulum. Enlargement of the lacrimal gland may be symmetric and extensive. Nasal and sinus involvement is usually also evident. | Systemic disease primarily affecting the upper respiratory tract, lungs, and kidneys. Orbital involvement is present in ~20% of cases. Ocular manifestations include scleritis, episcleritis, uveitis, and retinal vasculitis. |
Erdheim–Chester disease | In Erdheim–Chester disease with orbital involvement, there may be infiltration of the anterior compartment (preseptal space) and/or posterior compartment of the orbit with intra- and extraconal enhancing lesions replacing the orbital fat and encasing the optic nerve and the extraocular muscles. Involvement of the ocular adnexa includes lacrimal gland enlargement. | Rare idiopathic systemic histiocytic disorder of adults characterized by xanthogranulomatous infiltrates with lipid-laden histiocytes and Touton giant cells in the long tubular bones, skin, soft tissue, heart, pericardium, blood vessels, lung, pleura, retroperitoneum, kidneys, hypothalamic/pituitary axis (diabetes insipidus), and dura. Orbital involvement is very rare and tends to be bilateral. These adult patients present with progressive proptosis, ophthalmoplegia, and visual loss. |
Endocrine lesions | ||
Thyroid-associated orbitopathy (Graves disease) | Characteristic CT findings are exophthalmos accompanied by enlargement of extraocular muscles with sparing of the tendinous attachments to the globe. The inferior recti, followed by medial and superior rectus muscles, are most often and most severely affected, but any muscle may be affected. About 90% of patients have bilateral muscle involvement, in 5% one isolated muscle belly is involved. There may be focal low-density areas within the muscle bellies, but the muscle margins remain sharp. The retroglobular fat is increased in volume, even in the absence of muscle enlargement, and cause anterior bulging of the orbital septum, prolapse of an enlarged lacrimal gland, and stretching and straightening of the optic nerve. Fatty replacement and a stringlike appearance of the extraocular muscles are findings of a later, chronic, noncongestive phase. | Autoimmune orbital inflammatory condition associated with thyroid dysfunction. However, 10% of patients have neither clinical nor laboratory evidence of hyper-thyroidism (euthyroid ophthalmopathy). Most common cause of uni- or bilateral painless exophthalmos in young and middle-aged adults with female preference (M:F = 1:3–6). Visual loss from compressive and/or ischemic optic neuropathy results from increased extraocular muscle size and often massive increase in orbital fat. |
Vascular lesions | ||
Orbital venous lymphatic malformation (lymphangioma) | Lobulated, poorly circumscribed, multicystic hypodense mass. The attenuation may change after infection or intralesional bleeding. Presence of blood products with fluid–fluid levels is highly suggestive. Bony remodeling may be present with large lesions. Punctuate calcification, or phleboliths, are uncommon. Usually exhibit cyst wall rim enhancement, but solid enhancement of venous components can also occur. | Orbital venous lymphatic malformation is a congenital hamartomatous lymphatic and venous vascular anomaly. Orbital venous lymphatic malformations tend to populate the extraconal space but may be located superficial (conjunctiva or lid), deep (retrobulbar), and often transspatial. They gradually and progressively enlarge during the growing years and do not involute (unlike capillary hemangioma). Occurs in children and young adults, with slight female predominance. The tendency toward spontaneous hemorrhage results in the sudden onset of proptosis, periorbital swelling, reduced eye mobility, and optic nerve compression. |
Venous vascular malformation (cavernous hemangioma) | Unilateral, very rarely bilateral or multiple. Most commonly retrobulbar in the superior and lateral quadrants of the cone (80%). Appears as sharply circumscribed, rounded, ovoid, or lobulated homogeneous soft tissue mass of increased density. Calcified phleboliths are a nearly pathognomonic sign. Relatively uniform intense enhancement is the rule. The optic nerve is displaced by the tumor. Benign remodeling of bone in large lesions is not uncommon. | Cavernous hemangioma is a nonneoplastic, slow- growing hamartomatous vascular mass (low-flow lesion). It is the most common isolated orbital mass in adults 10 to 60 y of age (mean 40 y), with female predominance. Unilateral, slowly progressive, painless proptosis with diplopia and diminution of vision resulting from optic nerve compression is the usual presenting sign. Will not regress with age. |
Orbital varix | Uni- or bilateral dilation of one or more orbital veins, seen as round or elongated, tubular, sharply marginated, intra- or extraconal structures, often superolateral, that enhances intensely with contrast. Phleboliths may be associated. Enlarges in a prone position and during a Valsalva maneuver. Proptosis may be present, which may also be enhanced during a Valsalva maneuver. Thrombosis in varicoid veins may show a primarily hyperdense, dilated vein. After intravenous administration of iodinated contrast medium the thickened vessel wall may enhance intensely, contrary to the nonenhancing luminal thrombus. | Orbital varix is an intraorbital mass composed of abnormally large veins. It may be a single vessel with saccular or segmental dilation or a tangled plexus of venous channels. The enormous dilation is either of congenital (low-flow venous malformation) or acquired origin. The lesion may also occur in association with intraorbital or intracranial AVM. May present at any age with intermittent proptosis, elicited by hanging head, jugular tourniquet, Valsalva maneuver, or coughing. A varix is a leading cause of spontaneous orbital hemorrhage. Thrombosis is common. |
Carotid-cavernous sinus fistula | On imaging, proptosis combined with ipsilateral widening of cavernous sinus, inferior petrosal sinus, pterygoid venous plexus, and superior ophthalmic vein and enlargement of extraocular muscles is indicative of carotid-cavernous fistula. Choroidal effusions can be associated. Occasionally, sellar erosion and enlargement of the superior orbital fissure are also evident. Associated findings may include partial venous thrombosis in the lumen of the superior ophthalmic vein or cavernous sinus. If trauma was the etiology, CT shows associated skull base fractures and intracranial complications. | Direct fast-flow carotid-cavernous sinus fistulas are spontaneous (primarily in diabetic older women and patients with osteogenesis imperfecta or Ehlers–Danlos syndrome) or posttraumatic arteriovenous communications between the cavernous carotid artery and cavernous sinus. Indirect lower-flow carotid-cavernous sinus fistulas, or dural AVMs, are multiple small shunts between the meningeal or dural branches of both the internal and external carotid artery and the dural veins around the cavernous sinus. Both types of carotid-cavernous sinus fistula may present with chemosis and pulsatile proptosis associated with an orbital bruit on auscultation. Palsies of CN III, IV, and VI may occur. Severe secondary glaucoma may lead to rapid monocular blindness. |
Superior ophthalmic vein thrombosis | Enhancing walls and irregularity or absence of luminal enhancement of the dilated superior ophthalmic vein indicate partial or complete thrombosis. | Often associated with cavernous sinus thrombosis that is frequently a late complication of advanced paranasal sinus infection. Presents with ophthalmoplegia, proptosis, and chemosis. |
Benign neoplasms | ||
Infantile hemangioma (capillary hemangioma) | The capillary hemangiomas may be primary to the orbit or primarily cutaneous involving the periocular tissues and lid with secondary orbital extension. The capillary hemangiomas in the subcutaneous space may form well-defined masses, but usually the lesions have a nodular, irregular margin. When there is no cutaneous component, and the lesion is deep in the orbit, it has a tendency to involve the superomedial portion of the orbit, but extensive ones infiltrate most of the orbit and may extend intracranially. CT usually shows a lobulated, irregularly marginated, infiltrative, heterogeneous, extra- and intraconal soft tissue mass, slightly hypodense, with intense, homogeneous enhancement. | Hemangiomas of infancy are high-flow neoplastic tumors. They grow rapidly during the first year of life and usually regress spontaneously by age 5 to 10. Although the tumor prefers the facial structures and the cutaneous portion of the eyelid, expansion may be seen in any orbital compartment. More common in girls. Presents with proptosis as well as eyelid and conjunctiva swelling. |
Hemangiopericytoma | Spherical soft tissue mass with intense enhancement. In contrast to a cavernous hemangioma, the margins of the expansile mass are less well defined. Infiltration of muscles and erosion of the orbital bone may be present. Calcification is not a feature. | Uncommon vascular tumor that can arise wherever capillaries are present. Hemangiopericytomas may originate in the orbit or from the sinonasal cavities and then invade the orbit. They are aggressive lesions that can infiltrate the orbit in a multicompartmental fashion. About 50% of the cases are malignant. They become clinically manifest as progressing prop-tosis or lid swelling. There is no gender predominance. Mean age of presentation is the fourth decade. Hemangiopericytomas may be difficult to differentiate from other rare vasculogenic tumors, such as angioleiomyoma, malignant hemangioendothelioma, and fibrous histiocytoma. |
Neurofibroma | Localized or circumscribed neurofibromas present as a well-circumscribed, oval or fusiform, homogeneous tumor, more common in the superior quadrants, with density similar to brain, that enhances well with contrast. Plexiform neurofibromas present as elongated cords and nodules, poorly defined, irregular, and infiltrating, sometimes suggesting the image of a “bag of worms.” Diffuse neurofibromas appear as a well-enhancing, ill-defined mass with extension into surrounding tissues (resulting in enlargement of extraocular muscles). The CT appearance of malignant nerve sheath tumors is similar to nonmalignant tumors with the exception of bone destruction. | Localized neurofibromas, unrelated to any syndrome, are seen in middle-aged patients and present as a painless mass leading to nonaxial proptosis. Plexiform neurofibromas are seen in the first decade of life. Involvement of the eyelid is considered to be virtually pathognomonic for von Recklinghausen disease. Neurofibromatosis type 1 can also be found in patients with multiple neurofibromas. Neurofibromas have a low but real malignant potential. |
Schwannoma | Well-circumscribed, ovoid or fusiform, intraconal, heterogeneous mass with density similar to brain that enhances well with contrast. Low-density areas may represent cystic, necrotic, or hemorrhagic degeneration, regions of Antoni B cells or Antoni A cells intermixed with lipid-rich Schwann cells. The optic nerve may be compressed in lesions arising in the orbital apex, or it may be engulfed by the tumor. | Benign, slow-growing tumors of adults. Arise from the Schwann cells of the various nerve sheaths and constitute 1% to 8% of all orbital tumors. Branches of CN III, IV, V, and VI, sympathetic fibers, parasympathetic fibers, and the ciliary ganglion may be sources. The optic nerve is never the site of origin because it does not have Schwann cells. Usually an isolated lesion, 1% to 18% may be associated with neurofibromatosis type 2. Patients present with slowly progressive, painless proptosis, diplopia, and strabismus. Papilledema and optic atrophy with visual loss may be present with optic nerve compression. |
Paraganglioma | Intraconal, retrobulbar, well-circumscribed, homogeneous, uniformly enhancing soft tissue mass lesion, is generally attached to an extraocular muscle and separate from the optic nerve. Prominent orbital veins may be present. | Paragangliomas of the orbit are extremely rare. The site of origin is thought to be the ciliary nerve, ciliary ganglion, or orbital neural tissue. Multiple paragangliomas can occur in 10% to 20% of patients. Paraganglioma may occur simultaneously in the orbit and carotid body. Present at any age, especially over age 50 y, without any gender bias, with progressive prop-tosis, eye motility restriction, conjunctival congestion, diplopia, papilledema, pain, and decreased vision. Five to 10% of all paragangliomas are malignant with local invasion and metastases. |
Rosai–Dorfman disease | CT demonstrates large, heterogeneously enhancing, infiltrative soft tissue masses with striking proptosis. Fatty infiltration within the orbital mass is occasionally seen. Some cases have eyelid, lacrimal gland, and orbital involvement with extensive pre- and postseptal masses, but lesions can be purely retrobulbar with flattening of the globe. The optic nerve can be stretched and bowed by the abnormal orbital soft tissue. Sclerotic bone changes of the orbital wall are seen with sinonasal and intracranial extension. | Sinus histiocytosis with massive lymphadenopathy is a rare benign, idiopathic, proliferative disease of phagocytic histiocytes. Predominantly, it affects children and young adults, with slight male predominance. Most patients have painless bilateral cervical, axillary, and inguinal lymphadenopathy. Extranodal involvement includes the respiratory tract, skin, nasal cavity, intra-cranium, and skeletal system. Orbital involvement with proptosis, limited eye movement, eyelid edema, epiphora, and decreased visual acuity is usually unilateral. |
Malignant neoplasms | ||
Rhabdomyosarcoma | CT features include a well-defined intraconal and/or extraconal mass, isodense with the extraocular muscles, which becomes increasingly heterogeneous as it enlarges and with focal hemorrhage. The lesion demonstrates moderate to marked enhancement (dynamic CT reveals a low peak and slow washout on the density vs time curve). Invasion of the eyelid, sinuses, and base of the skull with associated bone destruction or remodeling is common. | Most common primary orbital malignancy in children and most common soft tissue malignancy of childhood, arising from pluripotential mesenchymal elements in the orbit or extraocular muscles. The peak incidence is in the first decade (average age 7–8 y). The characteristic clinical presentation is the rapid development of a painless unilateral proptosis in a child. |
Lymphoproliferative lesions of the orbit | Bilateral orbital involvement is relatively common. In retrobulbar involvement the lymphoid tumor infiltrates and replaces the orbital fat by perineural and perivascular spread. The involvement may be diffuse and poorly defined but may produce an isolated, well-circumscribed and lobulated mass with irregular margins, isodense to extraocular muscles with moderate homogeneous enhancement that typically molds around existing orbital structures, such as the globe. There is no indentation of the globe. Infiltration and thickening of the extraocular muscles are not common. Lymphoma may encase the optic nerve, occasionally simulating meningioma or orbital pseudotumor. | Lymphoid tumors represent 10% to 15% of orbital masses. Usually non–Hodgkin lymphoma, they compromise a wide spectrum of proliferative disorders ranging from malignant lymphoma to the benign pseudolymphoma to the reactive hyperplasia. Orbital lymphoma typically occurs in the 45- to 60-yold group. The presenting complaints are subacute development and progression of painless swelling, proptosis, and diplopia. It is not uncommon for orbital involvement to be the initial site of clinical presentation of lymphoma. |
Orbital leukemia | CT findings are usually nonspecific. Diffuse thickening of the sclera or uvea, proptosis with infiltration of the retrobulbar fat, similar to orbital pseudotumor, Graves disease, or orbital cellulitis, or more circumscribed involvement of the optic nerve–sheath complex, and invasion of the extraocular muscles may be seen. Extraconal subperiosteal involvement is the result of direct infiltration of orbital bone or soft tissue by leukemic cells. | Orbital leukemia may be seen in adults with chronic lymphocytic leukemia. In children with acute myelogenous leukemia, the orbital infiltration may form a mass lesion, referred to as granulocytic sarcoma or chloroma. |
Metastases | Intraconal metastatic lesions appear as a discrete infiltrating mass or diffuse lesion isodense to extraocular muscles and vascular structures. Mild to moderate contrast enhancement is noted. Metastatic scirrhous carcinoma of the breast may produce diffuse intraconal involvement with enophthalmos. Metastatic disease of the extraocular muscles may produce an asymmetric nodular configuration of the involved muscle or a diffuse muscle enlargement. When present, bony involvement may be osteolytic, osteoblastic, or both. | Metastases account for ~10% of orbital neoplasmas. Nearly all systematic malignancies have been reported to metastasize to the orbit (the most common source is, in descending order of frequency, breast and lung cancer, unknown primary, prostate, and melanoma; childhood tumors reported to metastasize to the orbit include neuroblastoma, Ewing sarcoma, Wilms tumor, and medulloblastoma). Metastases to fat and bone occur twice as frequently as that of extraocular muscles. Sixty percent of lesions are extraconal, 20% intraconal. With time, most metastases produce diffuse orbital infiltration. Patients with orbital metastases frequently complain of diplopia, ptosis, proptosis, eyelid swelling, pain, and vision loss. |










Disease | CT Findings | Comments |
Congenital/developmental lesions | ||
Naso-orbital cephalocele | When present, there is a defect in the bony confines, and CSF, meninges, or brain may show mass effect on the orbital structures. | Cephaloceles are extracranial herniations of intracranial contents due to partial failure of rostral neural tube closure. In naso-orbital cephaloceles, the defect occurs between the ethmoid and frontal processes of the maxilla and may pass into the orbit, appearing as a medial orbital mass. |
Dermoid | The superficial (simple, exophytic) subtype appears as a well-demarcated, ovoid mass, heterogeneous, cystic, with mixed fat and soft tissue density contents and mild, thin, rim enhancement, usually located extraconally in the superolateral aspect of the anterior orbit at the frontozygomatic suture, near the lacrimal gland, or in the superonasal aspect at the frontolacrimal suture. May contain fine or punctate calcifications in the cyst wall. Expansion or pressure erosions of the bony orbit may be associated. The deep (complicated, endophytic) subtype of adults may extend into sinuses, high deep masticator space, or intracranially, and cause a dumbbell-shaped dermoid. Ruptured orbital dermoid may be associated with irregular margins and reactive inflammatory changes indistinguishable from cellulitis. | The orbital ectodermal inclusion cysts are the most common congenital lesions of the orbit. Dermoids consist of epithelial elements plus dermal substructures including dermal appendages (e.g., hair or sweat glands in the wall). They most commonly present as superficial periorbital lesions in early childhood with proptosis. Most frequently presents in childhood and teenage years with painless subcutaneous mass, fixed to underlying bone, and slowly progressing, eccentric proptosis or progressive upper lid swelling. If ruptured, sudden growth, irregular margins, and inflammatory reactions may be present. Epidermoids are less common and can be differentiated from dermoids by features similar to fluid and more homogeneous appearance. Epidermoids contain epithelial elements only; do not contain fat. |
Orbital lymphatic-venous malformation | Lobulated, irregular, multicystic, hypodense mass with variable wall enhancement. Venous components may show a more diffuse enhancement. Layering hyperdense fluid-fluid levels correspond to blood products from spontaneous hemorrhage. Expansion or pressure erosions of the bony orbit may be associated. Calcifications or phleboliths are uncommon. | Congenital hamartomatous lymphatic and venous vascular malformation; orbital lymphangiomas tend to populate the extraconal space but are often trans-spatial. Lymphangiomas gradually and progresssively enlarge during the growing years (do not involute). The multilocular cystic lesions have a tendency to bleed. Orbital lymphatic-venous malformations occur in children and young adults, with slight female predominance. Most common clinical sign is progressive, painless proptosis with intermittent worsening. Optic nerve compromise is a late complication of large lesions. |
Inflammatory/infectious conditions | ||
Orbital subperiosteal abscess | Lentiform, low-density fluid collection running parallel to the wall of the orbit with adjacent sinus opacification and with central displacement of the extraocular muscle. Rarely, one may see a rim enhancement in this location or a small collection of gas. Demineralization, thinning, or loss of bone manifests inflammation or localized osteomyelitis of the adjacent bone. Cellulitic changes with stranding and streaking of the intraconal fat, enhancing and swollen extraocular muscles, or solid enhancing phlegmon retrobulbar may precede or follow abscess formation. Progression may lead to intraorbital abscess, superior ophthalmic vein thrombosis, cavernous sinus thrombosis, and intracranial extension. | Accumulation of pus between (usually medial) orbital wall and orbital periosteum, secondary to acute sinusitis and phlebitic transmission, or direct extension through dehiscence in the adjacent orbital bone (lamina papyracea). Uncommonly, due to trauma, bacteremia, or skin infection. Patients present with edema of the lids, painful proptosis, chemosis, eye muscle paralysis, visual disturbance, sinusitis, upper respiratory infection, and headache, fever, and chills. |
Benign neoplasms | ||
Meningioma | Sphenoid wing meningioma is the most common meningioma in the extraconal area. May involve the extraconal space from above or either side. A slightly hyperdense mass, with or without calcifications, and intense and uniform contrast enhancement is seen with sclerotic or lytic involvement of the adjacent orbital bone. | Extraconal meningiomas from the orbital wall periosteum or randomly located arachnoidal nests are infrequent. Meningiomas can involve the dura and contiguous bone of the orbital roof and greater wing of the sphenoid. Lesions can also involve the roof of the ethmoid or planum sphenoidale and can involve the sphenoid in the environs of the optic canal. Secondary extension into the orbit from intracranial meningiomas may also occur. Present most often in the fourth or fifth decade, occurring twice as often in females. |
Ossifying fibroma | Well-circumscribed, expansile, mineralized mass, surrounded by a thick or thin radiodense rimming. There may be islands of bone formation within the lesion. Periosteal reaction is not a feature of benign fibroosseous lesions. Enhancement is more pronounced in areas that are less mineralized. | Ossifying fibroma of the orbit and paraorbital region is often diagnosed incidentally, predilects to (black) women. Tends to occur in the third and fourth decades of life. |
Psammomatoid active ossifying fibroma | Expansile mass with admixture of both soft tissue and bone density pattern. There may be areas of soft tissue–fluidlike level. The most characteristic feature is the presence of numerous round or oval calcified bodies of various sizes, representing the psammomatoid (cementicle) bodies. The lesion has an aggressive appearance with cortical break and extension into surrounding tissue. | Psammomatoid active ossifying fibroma typically occurs in the sinonasal tract (ethmoid sinus, supraorbital frontal region) and potentially may behave aggressively with locally invasive and destructive capabilities. Occurs in younger age groups (first and second decades), with no gender predilection. |
Osteoma | Well-circumscribed, sharply delineated, round intrasinus mass, attached to the sinus wall broad-based or by a short pedicle, usually < 2 cm in size. Ivory osteomas are denser than adjacent bone; mature osteomas are isodense. Fibrous osteomas are lower and more variable in density. Most frequently seen in the frontal (80%) and ethmoid sinuses. Rarely, large osteoma in the frontal or ethmoid region may displace globe forward and cause proptosis. Obstruction of a sinus ostium may lead to infection or formation of a mucocele. Very rarely, an osteoma may erode through the dura, leading to rhinorrhea or intracranial infection. | Most common tumor of the paranasal sinuses. Osteomas are benign mature bone-forming hamartomatous lesions that are almost exclusively identified in the craniofacial skeleton (most common in the frontal and ethmoid sinuses). Symptoms associated with paraorbital osteomas include headaches, facial swelling or deformity, and proptosis and ocular disturbances. Paraorbital osteomas are more common in men, most often in the second to fourth decades of life. May be associated with Gardner syndrome. |
Osteoblastoma | Well-defined, round, expansile, mineralized lesion with prominent calcified rim (the central portion may have a similar appearance as ossifying fibroma). | Uncommon benign osteoblastic neoplasm. Head sites of involvement include the mandible, maxilla, temporal bone, paranasal sinuses, and orbit. Occurs more often in men younger than 30 y of age. Associated symptoms include pain, facial swelling, and asymmetry. |
Giant cell tumor (osteoclastoma) | Lobulated, expansile, intraosseous, mildly hyperdense soft tissue mass with marked homogeneous or heterogeneous contrast enhancement. Overlying thinned cortical shell, focally interrupted, often sclerotic. | Giant cell tumors are benign but locally aggressive neoplasms. Giant cell tumors of the head and neck are uncommon (sites of occurrence include the sphenoid, temporal, and ethmoid bones). Symptoms of lesions of the paraorbital region include headache, diplopia, decreased vision, and proptosis. |
Chondroma | Well-circumscribed lesion with endosteal scalloping composed of hyaline-type cartilage with varying degrees of calcification. | Chondromas of the paraorbital region, including the sinonasal tract (nasal cavity septum, ethmoid sinus) and nasopharynx are rare. There is equal gender predilection. Most patients are younger than 50 y of age. Symptomatic patients may present with nasal obstruction, enlarging, painless mass, proptosis, and headaches. Other, uncommon benign osseous neoplasms in the orbital region: chondroblastoma and chondromyxoid fibroma. |
Malignant neoplasms | ||
Osteosarcoma | Destructive, poorly delineated osteolytic, osteosclerotic, or mixed mass lesion, with minimal or massive tumor bone formation within the tumor proper and invading surrounding tissue. Subperiosteal tumor bone formation and displaced periosteum are characteristic. | Craniofacial osteosarcomas occur in the jaws, paranasal sinuses, skull, and orbital region. They have an equal gender predilection and occur generally a decade or two later than those with extrafacial osteosarcomas. May be seen following radiation to the orbit for retinoblastoma. |
Chondrosarcoma | May be seen as a nondestructive, fairly well delineated or destructive, and poorly delineated osteolytic or mixed lesion with coarse calcifications. There may be moderate to significant enhancement. | Craniofacial chondrosarcomas are slightly more common in men than in women and primarily occur in the fourth to seventh decades of life. An expanding mass may cause pain, proptosis, and visual disturbances. |
Rhabdomyosarcoma | Extraconal and/or intraconal soft tissue mass commonly associated with bone destruction and possible extraorbital extension into the eyelid, adjacent sinus, and intracranial cavity. The tumor is isodense to muscle and may enhance moderately to markedly. Tumors with focal hemorrhage appear heterogeneous. | The most common primary malignant tumor of the orbit (excluding the globe) in children. About 90% occur before age 16. Patients present with rapidly progressive unilateral proptosis, ptosis, palpable mass, and ophthalmoplegia. |
Metastases | Metastasis to the bony orbit presents on CT as bone destruction or hyperostosis. A soft tissue mass extending from the abnormal bone into the extraconal space or outside the orbit is often seen. | Metastatic disease to the bony orbit most commonly occurs in the greater sphenoid wing and may be limited to the bone or may have associated soft tissue components in the extraconal space or in the contiguous extraorbital location. |
Secondary orbital tumors | A low-density or soft tissue mass extending from outside the orbit into the extraconal space, sharply or poorly marginated, with or without contrast enhancement, with remodeling and bowing or with bony erosion and destruction of bony walls. The involvement of the orbit represents an advanced stage. | Malignant and benign tumors, including cysts, of the sinonasal cavities may invade the orbit. Tumors of the skin of the face can invade the orbit. Orbital invasion of intracranial tumors through the skull base may be present, either through natural foramina or by bone destruction. Malignant neoplasms of the oral cavity can extend along the perineural-perivascular pathway into the pterygopalatine fossa and then into the orbit. |
Metabolic/dysplastic lesions | ||
Fibrous dysplasia | Involvement by fibrous dysplasia is usually unilateral, which leads to asymmetry. Expansion of involved bone with a homogeneous or heterogenous decreased (“ground glass”) bone density, along with an intact thin cortex is typical, with an abrupt transition zone between the lesion and normal bone. May have cystic regions in the early, active phase of the disease, with centrally lucent lesions and thinned but sclerotic borders. The pagetoid (mixed) pattern of fibrous dysplasia shows mixed radiopacity and radiolucency. Obstructs osseous canals, foramina, pneumatic system, and sinuses. Contrast enhancement is often difficult to appreciate except in areas of lucent bone. | Fibrous dysplasia is a benign developmental skeletal disorder typically seen in adolescents and young adults younger than age 30. Can be monostotic or polyostotic. Orbital involvement may be focal or diffuse. Patients usually complain of diplopia and proptosis. Patients with orbitofacial neurofibromatosis type 1 may show sphenoid dysplasia with bony defects, middle fossa arachnoid cyst, optic nerve glioma, buphthalmos, and multiple plexiform neurofibromas. Often these patients present with pulsatile proptosis. |
Paget disease | Thickened bone with both lytic and sclerotic areas. When involved, the roof and lateral orbital walls are the most frequently diseased areas. | Involvement of the bony orbit by Paget disease is infrequent, occurring only late in the disease process. |
Trauma | ||
Orbital subperiosteal hematoma | The CT appearance of acute subperiosteal hematoma is a sharply defined, extraconal, homogeneous, high-density, nonenhancing, fusiform or biconvex mass. The muscles may be displaced inward by the extraconal collection. A swelling of the muscle and intraconal compartment may be associated. The CT appearance of chronic subperiosteal hematoma (hematic cyst) includes a well-defined extraconal, homogeneous or nonhomogeneous, nonenhancing mass, hyperdense (related to the protein-rich fluid and hemosiderin deposition) with osseous remodeling (erosion, expansion) in the majority of lesions. | Traumatic lesions of the orbital wall may form extraconal hematomas, usually along the roof or the floor and medial wall in association with a fracture. Nontraumatic acute and chronic subperiosteal hemorrhages of the orbit are rare and result from disruption of orbital vessels that penetrate the periosteum adjacent to the bone in association with sudden elevation of cranial venous pressure, bleeding diathesis, and paranasal sinusitis. They may present as orbital masses and may cause unilateral proptosis, limitation of eye mobility, and compressive optic neuropathy. The roof is the most common location; these hemorrhages occur almost exclusively in children and young adults (because the periosteal attachment to the roof tends to become stronger with age). Postoperative subperiosteal hematoma is a rare but severe complication after sinonasal operation. |
Orbital fractures | Direct fracture signs are alterations in skeletal contour and alignment, a linear lucency with irregular borders representing fracture line, traversing the orbital wall, with or without separation and displacement of the fragments, and changes in adjacent soft tissues. Indirect fracture signs, such as incorrectly located air (subcutaneous air inclusion, orbital emphysema, and pneumocephalus) or soft tissue densities (air-fluid levels in the sinus; the presence of a polypoid mass (tear-drop) representing herniated and entrapped fibrofatty tissues and/or muscles between bone fragments; and a small local hematoma (“hanging drop” at the fracture site) can provide additional diagnostic assistance. Orbital floor fractures can occur as isolated injuries or in combination with zygomatic arch fractures, Le Fort type II or III midface fractures, and medial wall or orbital rim fractures. Blow-out fractures commonly represent isolated fractures of the orbital floor without involvement of the rim with outward displacement of the dislocated fragment, fat herniation into the maxillary sinus, and inferior rectus muscle herniation into the maxillary sinus, resulting in muscle entrapment. Trapdoor fractures are pure internal orbital fractures. The anteroposterior linear fracture is hinged medially by a greenstick fracture. It involves the orbital floor, medial to or within the infraorbital groove and canal or medial wall. Entrapment of orbital tissue commonly occurs; may incarcerate herniated tissue. Medial blow-out fractures may be isolated but are more common in conjunction with inferior blow-out fractures. Isolated orbital rim, orbital roof, and lateral orbital wall fractures are relatively uncommon. Fracture of the zygomaticomaxillary complex (the term tripod fracture is misleading ) extends through the four articulations of the zygomatic bone: the zygomaticofrontal, the zygomaticosphenoidal, the zygomaticotemporal, and the zygomaticomaxillary sutures, with displacement and rotation of the zygoma. Additionally, this fracture may extend posteriorly to involve the pterygoid processes, greater sphenoid wing, and sphenotemporal buttress. Le Fort II fracture has a pyramidal shape and extends from the nasal bridge at or below the nasofrontal suture through the frontal processes of the maxilla, inferolaterally through the lacrimal bones and inferior orbital floor and rim through or near the infraorbital foramen, and inferiorly through the anterior wall of the maxillary sinus; it then travels under the zygoma, across the pterygomaxillary fissure, and through the pterygoid plates. Le Fort III fractures start at the nasofrontal and frontomaxillary sutures and extend posteriorly along the medial wall of the orbit through the nasolacrimal groove and ethmoid bones. The thicker sphenoid bone posteriorly usually prevents continuation of the fracture into the optic canal. Instead, the fracture continues along the floor of the orbit along the inferior orbital fissure and continues superolaterally through the lateral orbital wall, through the zygomaticofrontal junction and the zygomatic arch. Intranasally, a branch of the fracture extends through the base of the perpendicular plate of the ethmoid, through the vomer, and through the interface of the pterygoid plates to the base of the sphenoid. | Fractures of the orbital floor and medial wall occur with greater frequency than fractures of the roof, lateral wall, or complex orbital rim fractures. Isolated blow-in fractures of the four orbital walls are much less common than blow-up (superior blow-out) fractures or blow-out fractures of the orbital floor and the medial wall. |
| Orbital apex fractures are divided into (1) comminuted fractures, usually with fragment dislocation; (2) linear fractures, without dislocation of fragments; and (3) apex avulsion, with an intact optic canal; coexistent with orbital, facial, and skull base fractures. |
|
Miscellaneous lesions | ||
Langerhans cell histiocytosis | Osteolytic lesion, commonly in the superior or superotemporal orbit region with a fairly well-defined or diffuse soft tissue mass, with moderate to marked enhancement, encroaching lacrimal gland, lateral rectus, or even the globe. Multiple lesions may be present, resulting in multiple bony defects, on rare occasions in the orbital apex and superior orbital fissure. Similar lesions may be seen in facial bones, skull base, and calvarium. | Although Langerhans cell histiocytosis (LCH) is a rare disease, in patients with LCH, orbital involvement is not uncommon. Most of the patients are children 1 to 4 y old (age range birth-56 y). There is no gender predilection. Common symptoms of orbital LCH are unilateral or bilateral proptosis, edema, erythema of the eyelid, and periorbital pain, ptosis, optic nerve atrophy, and papilledema. |
Giant cell reparative granuloma | These soft tissue lesions enhance with contrast and present in two forms: a peripheral form, involving soft tissues (e.g., paraorbital, sinonasal, or oral), and a central form, confined to intraosseous sites. The central lesion causes osteolysis and may have a bubble-like appearance; it is usually well delineated and may contain calcifications. | Giant cell reparative granuloma is a benign reactive osseous proliferation. In the head and neck area, the maxilla and mandible are the most common sites of occurrence; orbital, paraorbital, or nasopharyngeal involvement is less common. They are more common in women (most younger than 30 y). Paraorbital involvement is associated with pain and swelling. |
Lacrimal gland pathology | ||
Inflammatory lesions of the lacrimal gland | ||
Idiopathic orbital inflammatory disease | Inflammation tends to cause diffuse, at times massive, enlargement of a lacrimal gland (neoplasms usually spare the palpebral lobe), with moderate to marked contrast enhancement. | Most common nonneoplastic lacrimal mass; presents with tenderness in the upper outer quadrant of the orbit in the region of the lacrimal gland. |
Dacryoadenitis | Contrast-enhanced CT typically demonstrates a massive contrast-enhancing lacrimal gland. Bilateral lacrimal masses are usually due to sarcoidosis (or lymphoproliferative lesions of the orbit). Abscess is diagnosed by identification of a characteristic low-density area with rim enhancement within an enlarged lacrimal gland. | Most acute, inflammatory enlargement of the lacrimal gland in younger patients results from postviral syndrome. Acute bacterial dacryoadenitis with suppuration and abscess formation is exceedingly rare and may develop secondary to an adjacent infection, from blood-borne spread or after trauma. Chronic dacryoadenitis, presenting with usually painless, slow lacrimal enlargement, may follow acute infection or is secondary to noninfectious inflammatory disorders such as sarcoidosis, Sjögren and Mikulicz syndrome, Kimura disease, and Wegener granulomatosis. |
Lacrimal gland neoplasms | ||
|
| Tumors of the lacrimal gland may be epithelial, lymphoid, or metastatic. They comprise 40% to 50% of all lacrimal masses, one half of which are benign mixed tumors, with the other half constituting malignant tumors. |
Benign mixed tumor (pleomorphic adenoma) | Well-circumscribed, round or oval, predominantly solid, partly cystic, partly calcified extraconal tumor, with moderate contrast enhancement, and smooth, scalloped remodeling of lacrimal fossa. Irregularity at the edge of the tumor or infiltration of the adjacent orbital tissue may be seen in malignant transformation. | Most common benign neoplasm of the lacrimal gland. Majority originates from the inner orbital lobe of the lacrimal gland. Clinical signs include a slow-growing, painless mass in the superolateral orbit of middle-aged patients (~40–50 y; without gender predilection), proptosis, and limited ocular motility. Other rare benign lacrimal gland tumors: oncocytoma, Whartin tumor. |
Adenoid cystic carcinoma | Extraconal, irregular, isodense, solid, homogeneous, diffuse enhancing mass arising within the lacrimal gland, with invasion of the adjacent bony orbit. Regional intracranial extension in advanced tumors. Calcifications are not uncommon. | Although uncommon, adenoid cystic carcinoma is the most frequent malignant tumor of the lacrimal gland. High incidence of perineural tumor spread (best visualized with MRI). Occurs in young adults to old age (peak fourth decade), without gender predilection. Clinical profile: rapid onset of symptoms, painful mass, pain with paresthesia, proptosis, ptosis, and limited ocular motility. Other rare malignant epithelial lacrimal gland tumors: ex pleomorphic or other adenocarcinomas, mucoepidermoid carcinoma, squamous cell carcinoma, and undifferentiated carcinoma. |
Lymphoproliferative lesions of the orbit | Homogeneous enhancing tumor, anywhere in orbit. Bilateral in 25%. Predilection for lacrimal gland (may be the only site): wedge-shaped enlargement of the lacrimal gland, isodense to slightly hyperdense, with moderate diffuse contrast enhancement. The extra-conal mass may present with anterior and/or posterior extension and molds to and encases normal orbital structure. Malignant variants may have infiltrative appearance. | Lymphoproliferative lesions of the orbit include a wide spectrum, ranging from reactive hyperplasia, low-grade primary small B-cell lymphoma (especially mucosa- associated lymphoid tissue [MALT]), to diffuse large B-cell lymphoma, Burkitt lymphoma, and T-cell lymphoma. They comprise 5% to 10% of orbital masses. Although rare, primary lymphomas without systemic involvement appear to be the most common nonepithelial tumor of the lacrimal gland. Extramedullary plasmacytoma may also be seen in the lacrimal gland (extremely rare). |
Metastases | Metastatic tumor in a lacrimal gland has a nonspecific CT appearance of diffuse enlargement of the gland. The lacrimal gland may be involved along with the adjacent periorbita and extraconal spaces. | Metastasis to the extraconal space may involve the lacrimal glands or the fat-containing spaces posterior to the orbital septa. |
Miscellaneous lesions of the lacrimal gland | ||
Focal amyloidosis | Amyloidoma of the lacrimal gland is associated with a unilateral lacrimal gland mass of soft tissue density, without contrast enhancement, frequently with punctate calcification, molding to surrounding orbital structures. | Focal amyloidosis is a very uncommon disorder. It may involve different orbital structures. Isolated involvement of the lacrimal gland may mimic inflammatory or even tumorous lesions. It typically affects middle-aged women with painless progressive proptosis but without visual morbidity. CT findings may mimic those seen in inflammatory or lymphoproliferative disorders of the lacrimal gland. |
Lacrimal intraglandular cyst (dacryops) | Lacrimal gland enlargement due to cystic structures with fluid density and thin peripheral contrast enhancement that are contiguous with the palpebral lobe of the lacrimal gland. Calcifications are uncommon. No bone erosion. | Obstruction of the lacrimal gland ductules of the major and accessory lacrimal glands leads to the formation of dacryops, ductal cysts of the lacrimal glands. Dacryops is a rare clinical phenomenon, most commonly presenting unilaterally in the palpebral lobe. Patients frequently complain of painless swelling in the lateral portion of the upper eyelid. A history of trauma or inflammation of the conjunctiva or a congenital anomaly of the excretory duct can be the precipitating factor to cyst formation. Benign ductal cysts of the accessory lacrimal glands are uncommon lesions of the orbit arising from the glands of Krause and Wolfering. |













Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


