Accurate diagnosis of Aβ pathology
Prognosis of conversion to AD
Assessment of the spatial and temporal pattern of Aβ deposition and its relation to disease progression, cognitive decline and other disease biomarkers
Early detection of Alzheimer’s disease pathology, allowing disease-specific interventions when cognition is unimpaired or minimally impaired
Subject selection for anti-Aβ trials
Predict response to anti-Aβ therapy
Monitor the effectiveness of anti-Aβ therapy
As a quantitative neuroimaging probe, the Aβ radiotracer must possess a number of key general properties: it should be a lipophilic, nontoxic small molecule with a high specificity and selectivity for Αβ, amenable for high specific activity labelling with 18F or other long lived radioisotope, with no radiolabelled metabolites that enter the brain, while reversibly binding to Αβ in a specific and selective fashion (Laruelle et al. 2003; Nordberg 2004; Pike 2009). Overall, binding affinity and lipophilicity are the most crucial properties for in vivo neuroimaging tracers. While high affinity for Αβ is desirable to provide an adequate signal-to-noise ratio, this high affinity might also delay reaching binding equilibrium requiring an extended scanning time. And while lipophilicity is necessary for the tracer to cross the blood-brain barrier (BBB), if the radiotracer is too lipophilic, its nonspecific binding might be too high (Laruelle et al. 2003; Pike 2009).
Through the years, several compounds have been evaluated with different fortune as potential Aβ probes: monoclonal and anti-Aβ antibodies fragments, serum amyloid P, basic fibroblast growth factor, Aβ fragments, derivatives of histopathological dyes such as Congo red, chrysamine G and thioflavin S and T (for review, see (Villemagne and Rowe 2010)). While selective tau imaging for in vivo NFT quantification is still in the early stages of development (Okamura et al. 2005; Maruyama et al. 2009; Ojida et al. 2009; Fodero-Tavoletti et al. 2011), several Αβ radiotracers found their way into human clinical trials.
Almost a decade after unsuccessful trials with anti-Αβ antibodies (Majocha et al. 1992), Αβ imaging came to fruition with the first report of successful imaging in an AD patient with 18F-FDDNP, a tracer claimed to bind both plaques and NFT (Shoghi-Jadid et al. 2002). Since then, human Αβ imaging studies have been conducted in AD patients, normal controls and patients with other dementias using 11C-PiB (Klunk et al. 2004), 11C-SB13 (Verhoeff et al. 2004), 11C-ST1859 (Bauer et al. 2006), 11C-BF-227 (Kudo et al. 2007), 11C-AZD2138 (Nyberg et al. 2009), 18F-florbetaben (Rowe et al. 2008), 18F-flutemetamol (Serdons et al. 2009a), 18F-florbetapir (Wong et al. 2010) and 18F-AZD4694 (Cselenyi et al. 2012) with PET.
In order to test compounds that could have more widespread application, preliminary studies with SPECT Aβ radiotracers 123I-CQ (Opazo et al. 2006) and 123I-IMPY (Newberg et al. 2006) showed limited utility for the evaluation of Aβ burden in AD, although 123I-IMPY might be useful in the evaluation of transmissible spongiform encephalopathies (Song et al. 2008). New SPECT radiotracers labelled with 123I or those that could be potentially labelled with 99mTc are being evaluated (Qu et al. 2007a, b; Serdons et al. 2007; Lin et al. 2009).
While all of the aforementioned tracers bind with varying degrees of success to Αβ fibrils and brain homogenates of AD patients, Congo red and thioflavin T—and some of their derivatives—have recently been shown to also bind to soluble oligomeric forms of Αβ (Maezawa et al. 2008). On the other hand, Αβ soluble species represent less than 1 % of the total brain Αβ (McLean et al. 1999), and the reported affinity of PiB for these soluble oligomers seems to be significantly lower than for Αβ fibrils (Maezawa et al. 2008). Until highly selective radiotracers are developed to bind the Αβ soluble species, the contribution of these oligomers to the PET signal in sporadic AD from tracers such as 11C-PiB is considered to be negligible (Mathis et al. 2007).
10.2.1 11C-Labelled Radiotracers
11C-PiB
11C-PiB (PiB), the most successful and widely used of the currently available Αβ tracers, has been shown to possess high affinity and high selectivity for fibrillar Αβ in plaques and other Αβ-containing lesions (Klunk et al. 2001; Mathis et al. 2002, 2005; Price et al. 2005; Ye et al. 2005; Cohen et al. 2012). In vitro studies with high specific activity 3H-PiB demonstrated two binding sites for PiB in brain homogenates and Aβ1–42 fibrils (Klunk et al. 2005). Recent studies have shown that 11C-PiB binds with high affinity to the N-terminally truncated and modified Αβ and AβN3-pyroglutamate species in senile plaques (Maeda et al. 2007). In vitro assessment of 3H-PiB binding to white matter homogenates failed to show any specific binding (Fodero-Tavoletti et al. 2009b). PiB is a derivative of thioflavin T, a fluorescent dye commonly used to assess fibrillisation into β-sheet conformation (LeVine 1999), and as such PiB has been shown to bind to a range of additional Αβ-containing lesions including diffuse plaques and cerebral amyloid angiopathy (CAA) (Lockhart et al. 2007), as well as to Αβ oligomers—although with lower affinity (Maezawa et al. 2008). PiB also displayed lower affinity towards other misfolded proteins with a similar β-sheet secondary structure such as α-synuclein (Fodero-Tavoletti et al. 2007; Ye et al. 2008) and tau (Lockhart et al. 2007; Ikonomovic et al. 2008). This is relevant particularly since AD has been described as a ‘triple brain amyloidosis’ (Trojanowski 2002). Most importantly, these studies have shown that, at the concentrations achieved during a PET scan, 11C-PiB cortical retention in AD or DLB primarily reflects Αβ-related cerebral amyloidosis and not binding to Lewy bodies (LB) or NFT (Klunk et al. 2003; Fodero-Tavoletti et al. 2007; Lockhart et al. 2007; Ikonomovic et al. 2008). 11C-PiB has consistently provided quantitative information on Αβ burden in vivo, contributing new insights into Αβ deposition in the brain, allowing earlier detection of AD pathology (Klunk et al. 2004; Mintun et al. 2006; Rowe et al. 2007; Cohen et al. 2012) and accurate differential diagnosis of the dementias (Ng et al. 2007b; Rabinovici et al. 2007; Rowe et al. 2007).
11C-BF-227
11C-BF-227 (BF-227), a benzoxazole derivative, is also a promising tool as an Aβ imaging tracer (Okamura et al. 2004). In vitro binding studies with BF-227 demonstrated a Ki value of 4.3 nM for the compound to Aβ1–42 fibrils (Kudo 2006). A PET study using BF-227 showed AD patients were clearly distinguishable from age-matched controls with AD patients displaying significantly higher tracer retention in the cerebral cortex than controls (Kudo et al. 2007). Regional parametric analysis of the images further demonstrated a higher BF-227 retention in the posterior association cortex in AD patients. Unlike PiB, BF-227 presents with similar binding affinities for Aβ and α-synuclein (Fodero-Tavoletti et al. 2009a).
A few other C-11 Aβ radiotracers have been tested in humans. The evaluation of the stilbene derivative, 11C-SB13, showed that it could differentiate between five AD and six controls but had a lower effect size values when compared with 11C-PiB (Verhoeff et al. 2004). A preliminary study with the anti-Aβ agent 11C-ST1859 revealed small differences in radiotracer retention between nine AD patients and three healthy controls, but showing that the biodistribution and specificity of therapeutic agents can be assessed with PET (Bauer et al. 2006). Another Aβ radiotracer, 11C-AZD2138, showed not only reversible binding but also displayed very low nonspecific binding to white matter (Johnson et al. 2009; Nyberg et al. 2009).
10.2.2 18F-Labelled Radiotracers
Unfortunately, the 20-min radioactive decay half-life of carbon-11 (11C) limits the use of 11C-PiB to centres with an on-site cyclotron and 11C radiochemistry expertise, making the cost of studies prohibitive for routine clinical use. To overcome these limitations, several Aβ tracers labelled with fluorine-18 (18F; half-life of 110 min) that permits centralised production and regional distribution have been developed and tested. Recent studies with the newly introduced 8F-labelled Aβ-specific radiotracers, 18F-florbetapir (Wong et al. 2010; Clark et al. 2011), 18F-florbetaben (Rowe et al. 2008; Barthel et al. 2011; Villemagne et al. 2011a), 18F-flutemetamol (Nelissen et al. 2009; Vandenberghe et al. 2010b) and 18F-AZD4694 (Cselenyi et al. 2012), have been successfully replicating the results obtained with 11C-PiB. The latest entry into the list of fluorinated Aβ radiotracers is 18F-MK-3328, developed by Merck, that has the disadvantage that >17 % of its cortical signal was attributed to binding to monoamine oxidase-B (MAO-B) (Sur et al. 2010).
18F-FDDNP
A marked progression in the development of Aβ imaging tracers was the synthesis and characterisation by Barrio and colleagues of a very lipophilic radiofluorinated 6-dialkylamino-2-naphthyethylidene derivative that presents nanomolar affinity to Aβ fibrils (Barrio et al. 1999; Agdeppa et al. 2001a, b). 18F-FDDNP is reported to bind both the extracellular Aβ plaques and the intracellular NFT in AD (Shoghi-Jadid et al. 2002) while also binding to prion plaques in Creutzfeldt-Jakob disease (CJD) brain tissue (Bresjanac et al. 2003). However, in vitro evaluation of FDDNP in concentrations similar to those achieved during a PET scan showed limited binding to both NFT and Aβ plaques (Thompson et al. 2009). 18F-FDDNP was used to obtain the first human PET images of Aβ in an 82-year-old woman with AD. AD patients present with higher accumulation and slower clearance of 18F-FDDNP than controls in brain areas such as the hippocampus (Shoghi-Jadid et al. 2002). Retention time of 18F-FDDNP in these brain regions was correlated with lower memory performance scores, regional glucose hypometabolism and brain atrophy (Small et al. 2002, 2006). However, the dynamic range of 18F-FDDNP cortical uptake values between HC and AD patients is small (9 %). Direct comparison of 18F-FDDNP with 11C-PIB in monkeys (Noda et al. 2008) and in human subjects showed very limited dynamic range of 18F-FDDNP (Shih et al. 1987; Tolboom et al. 2009b) and, in a longitudinal study, was found to be less useful than 11C-PiB and 18F-fluorodeoxyglucose (FDG) for examining disease progression (Ossenkoppele et al. 2012). Interestingly, 18F-FDDNP still remains the only Aβ tracer showing retention in the medial temporal cortex of AD patients (Shih et al. 1987) and correlation with cerebrospinal fluid (CSF) tau (Tolboom et al. 2009b).
18F-AZD4694
Developed by AstraZeneca, the most salient features of 18F-AZD4694 are, as with 11C-AZD2138, fast tracer kinetics and low nonspecific binding to white matter, similar to that one observed with 11C-PiB, suggesting it might be helpful for the detection of small Aβ cortical deposits at very early stages of the disease process (Sundgren-Andersson et al. 2009; Jureus et al. 2010). Initial clinical studies with 18F-AZD4694 showed a clear distinction in tracer retention between six healthy controls and ten AD patients (Cselenyi et al. 2012).
18F-Florbetaben
18F-Florbetaben (a.k.a. AV1; BAY94-9172), synthesised by Kung and colleagues (2005b) and developed by Bayer Healthcare, has been shown to bind with high affinity to Aβ in brain homogenates and selectively labelled Aβ plaques and CAA in AD tissue sections (Zhang et al. 2005b). After injection into Tg2576 transgenic mice, ex vivo brain sections showed localisation of 18F-florbetaben in regions with Aβ plaques as confirmed by thioflavin binding (Zhang et al. 2005a). At tracer concentrations achieved during human PET studies, florbetaben did not show binding to LB or NFT tau in postmortem cortices from DLB or FTLD patients (Fodero-Tavoletti et al. 2012). In human studies, cortical retention of 18F-florbetaben was higher at 90-min postinjection in all AD subjects compared to age-matched controls and FTLD patients, with binding matching the reported postmortem distribution of Aβ plaques (Rowe et al. 2008). Recently completed phase II clinical studies further confirmed these results (Barthel et al. 2011), and an ongoing longitudinal study in MCI subjects is helping to establish the usefulness of FBB PET in predicting progression to AD (Ong et al. 2010). 18F-Florbetaben is highly correlated with 11C-PiB (r = 0.97 with a slope of 0.71) (Villemagne et al. 2012c) and was used to detect the presence or absence of AD pathology in the brain in subjects from a wide spectrum of neurodegenerative diseases (Villemagne et al. 2011a). Phase III studies for 18F-florbetaben have reached completion (Sabbagh et al. 2012).
18F-Florbetapir
As 18F-florbetaben, 18F-florbetapir is a stilbene derivative that was also synthesised by Kung and colleagues at the University of Pennsylvania (Zhang et al. 2005a) and has been developed by Avid Radiopharmaceuticals. Initial in vitro evaluation showed binding to Aβ plaques in AD brain sections (Choi et al. 2009; Lin et al. 2009). The most salient feature of this tracer is its rapid reversible binding characteristics allowing scanning only at 45–50 min after injection, similar to 11C-PiB (Wong et al. 2010). 18F-Florbetapir (Lister-James et al. 2011) has become the most widely used Aβ radiotracer after 11C-PiB and has been adopted as the Aβ radiotracer for ADNI 2 and also used in patient selection and to evaluate treatment response in a large number of multicenter therapeutic trials around the world. Several multicenter phase I and II studies in AD, MCI and HC showed the ability of 18F-florbetapir to discriminate between AD and age-matched controls, with ~50 % of the MCI subjects presenting with high 18F-florbetapir retention (Fleisher et al. 2011). Multicenter studies using 18F-florbetapir to assess the relationship between Aβ burden and cognition showed that in clinically normal older individuals, Aβ burden in the brain is associated with poorer memory performance (Sperling et al. 2013), while MCI subjects with high Aβ burden in the brain are at a significantly higher risk of cognitive decline over 18 months (Doraiswamy et al. 2012). An initial phase III study in 35 volunteers demonstrated a 96 % agreement between 18F-florbetapir and neuropathology for the detection of brain Aβ in vivo and no retention in young controls (Clark et al. 2011). An extension phase III in 59 volunteers establishes that 18F-florbetapir has a sensitivity of 92 % and a specificity of 100 % for the detection of Aβ pathology (Clark et al. 2012). Using a semiquantitative approach, it has been shown that 18F-florbetapir is >90 % sensitive and specific to detect Aβ pathology in the brain (Camus et al. 2012) and that it is correlated with 11C-PiB (r = 0.77 with a slope of 0.33) (Wolk et al. 2012b). 18F-Florbetapir (as Amyvid®) is the first radiotracer approved by the Food and Drug Administration (FDA) for detection of Aβ in vivo and the first 18F-labelled radiotracer approved by the FDA since FDG.
18F-Flutemetamol
Another fluorinated tracer, developed by GE Healthcare, that also recently completed a phase III study (Buckley et al. 2012) is 18F-flutemetamol (a.k.a. GE067) (Serdons et al. 2009a, b). Phase I and II studies demonstrated that 18F-flutemetamol can differentiate between AD and HC (Nelissen et al. 2009; Vandenberghe et al. 2010b) and that when combined with brain atrophy it could be predictive of disease progression in MCI subjects (Thurfjell et al. 2012). 18F-Flutemetamol brain retention is also highly correlated with 11C-PIB (r = 0.91 with a slope of 0.90) (Vandenberghe et al. 2010b). Furthermore, there was a high correlation between Aβ burden as measured by 18F-flutemetamol and by immunohistochemical assessment of brain biopsy tissue (Wolk et al. 2011; Wong et al. 2013).
While the cortical retention of 18F-florbetapir, 18F-florbetaben and 18F-flutemetamol provides a clear separation of AD patients from HC subjects, the degree of cortical retention with 18F-florbetapir and 18F-florbetaben is lower than with 11C-PiB (Villemagne et al. 2012c; Wolk et al. 2012b), showing a narrower dynamic range of SUVR values, that visually appears as a relatively higher degree of nonspecific binding to white matter. While the cortical retention of 18F-flutemetamol is similar to that of 11C-PiB, the nonspecific retention in white matter is much higher (Vandenberghe et al. 2010b). Due to this lower signal-to-noise ratio, visual readouts of the images are more challenging than with 11C-PiB (Rowe and Villemagne 2011). While 11C-PiB PET images in subjects with AD pathology usually clearly show high radiotracer retention in the grey matter in excess of that in subjacent white matter, these novel 18F tracers frequently show loss of the normal grey-white matter demarcation as the predominant evidence of cortical Aβ deposition (Rowe and Villemagne 2011).
To date, the vast majority of the Aβ imaging literature is based on 11C-PiB studies, with studies using the first generation of fluorinated Aβ radiotracers only recently being reported. The following survey of the state of the field is a reflection of this, and, unless otherwise specified, the reported results are from 11C-PiB studies.
10.3 Aβ Imaging in Alzheimer’s Disease
On visual inspection, cortical 11C-PiB retention is higher in AD, with a regional brain distribution that is highest in frontal, cingulate, precuneus, striatum, parietal and lateral temporal cortices, while occipital, sensorimotor and mesial temporal cortices are much less affected (Fig. 10.1). Both quantitative and visual assessments of PET images present a pattern of radiotracer retention that seems to replicate the sequence of Αβ deposition found at autopsy (Braak and Braak 1997), with initial deposition in the orbitofrontal cortex, inferior temporal, cingulate gyrus and precuneus, followed by the remaining prefrontal cortex and lateral temporal and parietal cortices (Fig.10.2). Multimodality studies in early AD have shown that the regional pattern of Αβ deposition is similar to the anatomy of the ‘default network’ (Buckner et al. 2005; Sperling et al. 2009), a specific, anatomically defined brain system responsible for internal modes of cognition, such as self-reflection processes, conscious resting state or episodic memory retrieval (Buckner et al. 2008; Wermke et al. 2008), an association that is present even in non-demented subjects (Drzezga et al. 2011).
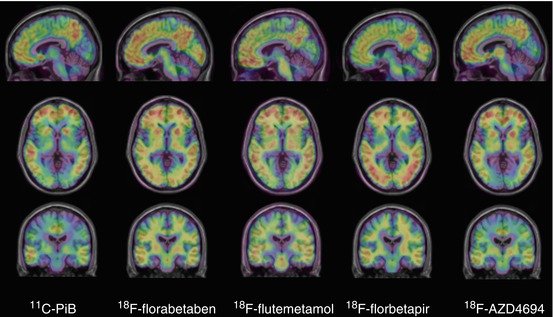
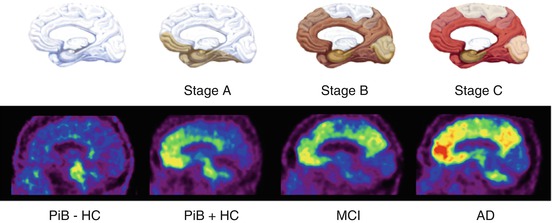
Fig. 10.1
Representative sagittal (top row), transaxial (middle row) and coronal (bottom row) PET images overlaid on MRI from Alzheimer’s disease patients obtained with different Aβ imaging radiotracers. From left to right, 11C-PiB, 18F-florbetaben, 18F-flutemetamol, 18F-florbetapir and 18F-AZD4694. The images show the typical pattern of tracer retention in AD, with the highest retention in frontal, temporal and posterior cingulate cortices, reflecting Aβ plaque burden
Fig. 10.2
Top row. Schematics showing the stages of Aβ deposition in the human brain as proposed by Braak and Braak (1997). Bottom row. Representative sagittal PET images showing the regional brain distribution of 11C-PIB in two asymptomatic healthy age-matched controls, one with low (PiB- HC) and one with high Aβ burden (PiB + HC), a subject classified as mild cognitive impairment (MCI) with significant Aβ deposition in the brain and an Alzheimer’s disease (AD) patient with even a higher Aβ burden in the brain. The pattern of PiB retention replicates the sequence of Αβ deposition described from postmortem studies (Braak and Braak 1997)
The regional distribution of 11C-PiB retention is not always the same and varies with the specific Αβ distribution characteristic of the underlying pathology. For example, carriers of mutations associated with familial AD (Klunk et al. 2007; Koivunen et al. 2008b; Villemagne et al. 2009a), subjects with posterior cortical atrophy (Ng et al. 2007b; Tenovuo et al. 2008; Kambe et al. 2010; Formaglio et al. 2011) or CAA (Johnson et al. 2007; Dierksen et al. 2010) each presenting a different regional 11C-PiB distribution of retention to the one usually observed in sporadic AD (Klunk et al. 2004; Rowe et al. 2007).
Quantitative PET studies using diverse Αβ radiotracers have shown a robust difference in retention between AD patients and age-matched controls (Klunk et al. 2004; Price et al. 2005; Mintun et al. 2006; Kudo et al. 2007; Pike et al. 2007; Rabinovici et al. 2007; Rowe et al. 2007, 2010; Jack et al. 2008a; Morris et al. 2009; Furst et al. 2012; Barthel et al. 2011; Fleisher et al. 2011; Villemagne et al. 2011a; Camus et al. 2012; Cselenyi et al. 2012).
Longitudinal studies with 11C-PiB are showing that significant, although small, increases in Αβ deposition can be measured but also that these increases in Αβ deposition are present in the whole spectrum of cognitive stages, from cognitively unimpaired individuals to AD dementia patients (Engler et al. 2006; Jack et al. 2009; Okello et al. 2009b; Resnick et al. 2010; Rinne et al. 2010; Sojkova et al. 2011b; Villemagne et al. 2011b; Villain et al. 2012). Αβ accumulation is observed in individuals considered to have low Αβ loads with about 7 % of them progressing above the threshold in about 2.5 years (Vlassenko et al. 2011). Αβ accumulation is a slow process, and the evidence suggests that it remains constant in the preclinical and prodromal stages of the disease in those individuals deemed to have high Αβ burdens while being slightly higher at the early stages of AD (Villemagne et al. 2011b). In individuals with the highest Aβ burdens independent of clinical classification and also as Alzheimer’s dementia progresses, the rates of Aβ deposition start to slow down, although not completely reaching a plateau (Villain et al. 2012). This slower rate of Αβ deposition associated with disease progression might be attributed to a saturation of the process or, more likely, extensive neuronal death that precludes Αβ production.
10.4 Antemortem and Postmortem Correlations
While clinicopathological studies have shown that the accuracy of clinical assessments for the diagnosis of AD ranges between 70 and 90 %, depending on the specialisation of the centre, the regional retention of 11C-PiB and the novel 18F radiotracers is highly correlated with regional Αβ plaques as reported at autopsy or biopsy (Bacskai et al. 2007; Ikonomovic et al. 2008; Leinonen et al. 2008b; Villemagne et al. 2009b; Burack et al. 2010; Clark et al. 2011; Kadir et al. 2011; Sabbagh et al. 2011; Sojkova et al. 2011a; Wolk et al. 2011; Wong et al. 2013), with a higher Aβ concentration in the frontal cortex than in hippocampus, consistent with previous reports (Arnold et al. 2000; Naslund et al. 2000). As mentioned before and in contrast with CSF studies, Αβ imaging studies can assess both the quantity and regional brain distribution of Αβ, as observed in the different patterns of regional distribution of Αβ observed in sporadic AD (Klunk et al. 2004; Nordberg 2007), familial AD (Klunk et al. 2007; Koivunen et al. 2008b; Villemagne et al. 2009a), familial British dementia (Villemagne et al. 2010) and CAA subjects (Johnson et al. 2007; Dierksen et al. 2010). The results from Αβ imaging might be considered for the brain regions to be sampled and stained for the neuropathological diagnosis of AD (Montine et al. 2012).
There have been a handful of cases reported with a discrepancy between a positive neuropathological finding or a strong biomarker profile of AD and a low PET signal. To date, only seven discrepant cases have been reported, three of them in mutation carriers (Tomiyama et al. 2008; Schöll et al. 2011) and four in sporadic cases (Leinonen et al. 2008a; Cairns et al. 2009; Sojkova et al. 2011a; Ikonomovic et al. 2012). No ‘false-positive’ cases have been reported. Tomiyama and colleagues reported a case of a novel APP mutation (E693Delta) where the mutant Αβ does not fibrillise in the same way as wild-type Aβ (Inayathullah and Teplow 2011) in which the 11C-PiB study showed low cortical retention (Tomiyama et al. 2008). Schöll and colleagues report on two subject carriers of the Arctic APP mutation (Schöll et al. 2011). These two mutation carriers, while presenting with low 11C-PiB retention, had a clear ‘AD pattern’ of brain hypometabolism as measured by FDG, low Aβ1–42 and elevated t-tau and p-tau in CSF (Schöll et al. 2011). While neuropathology was not available in these two subjects, a family member carrier of the same mutation presented with atypical plaque morphology—with ringlike plaques lacking a congophilic core—at autopsy (Basun et al. 2008). In regard to sporadic cases, Leinonen et al., while correlating 11C-PiB retention and frontal lobe Αβ burden from biopsies on subjects undergoing intraventricular monitoring for normal pressure hydrocephalus, described a non-demented case with plaques that had a negative 11C-PiB scan (Leinonen et al. 2008a). Cairns et al. reported a longitudinally characterised subject with a low 11C-PiB scan that presented with evidence of cognitive decline and a CSF profile indicative of AD a year later (Cairns et al. 2009). At the time of autopsy, two and a half years after the 11C-PiB PET, diffuse Αβ plaques were observed with only minimal neuritic plaques and NFT pathology. Sojkova et al. reported imaging-neuropathological correlations in five participants from the Baltimore Longitudinal Study of Aging (Sojkova et al. 2011a). While there was agreement in four subjects, even in one that had only sparse plaques at autopsy, a fifth presented with low global and regional 11C-PiB retention despite presenting moderate plaques at neuropathological examination and meeting CERAD criteria for ‘possible AD’ (Mirra et al. 1991). Ikonomovic et al. (2012) reported autopsy findings in a subject with a clinical diagnosis of DLB and possible AD that presented with low 11C-PiB retention 17 months before death. At autopsy, focally frequent neocortical neuritic plaques fulfilled CERAD criteria of ‘definite AD’ (Mirra et al. 1991). Cortical Αβ1–40 concentration levels were on the same range to those found in a typical AD case with high 11C-PiB retention, but Αβ1–42 concentrations were lower. Furthermore, the majority of plaques labelled weakly with 11C-PiB and 3H-PiB binding were low, all findings suggesting low fibrillar Αβ.
On the one hand, these cases illustrate the possibility that different conformations of Aβ deposits (Levine and Walker 2010b) may affect the binding of tracers and that Aβ imaging may not recognise all types of Aβ pathologies with equal sensitivity (Walker et al. 2008; Rosen et al. 2011). On the other, it raises issues not only in regard to the sensitivity of Aβ imaging (e.g. What is the minimal amount of Aβ detected by PET?) but also in regard to the evaluation of brain tissue (e.g. type of antibodies used for immunohistochemistry and ELISA, fluorescent dyes, diagnostic criteria).
10.5 Aβ Deposition in Non-Demented Individuals and Its Relation with Cognition
Αbout 25–35 % of elderly subjects performing within normal limits on cognitive tests present with high cortical 11C-PiB retention, predominantly in the prefrontal and posterior cingulate/precuneus regions (Mintun et al. 2006; Rowe et al. 2007, 2010; Aizenstein et al. 2008; Villemagne et al. 2008b; Mormino et al. 2009; Reiman et al. 2009b). These findings are in perfect agreement with postmortem reports that ~25 % of non-demented older individuals over the age of 75 have Aβ plaques (Davies et al. 1988; Morris and Price 2001; Forman et al. 2007). Aβ deposition in these non-demented individuals (Mintun et al. 2006; Rowe et al. 2007; Aizenstein et al. 2008; Villemagne et al. 2008b) might reflect preclinical AD (Price and Morris 1999). Furthermore, the prevalence of subjects with high 11C-PiB retention increases each decade at the same rate as the reported prevalence of plaques in non-demented subjects in autopsy studies (Rowe et al. 2010). While cross-sectional examination of these controls essentially shows no significant differences in cognition (Mintun et al. 2006; Aizenstein et al. 2008; Jack et al. 2008a; Villemagne et al. 2008b; Rowe et al. 2010), significant cognitive differences in females were reported (Pike et al. 2011). More recently, Sperling and colleagues reported that high Aβ deposition in the brain was correlated with lower immediate and delayed memory scores in otherwise healthy controls (Sperling et al. 2013). The detection of Aβ pathology at the presymptomatic stage is of crucial importance because if this group truly represents preclinical AD (Price and Morris 1999; Thal et al. 2004; Backman et al. 2005; Small et al. 2007), it is precisely the group that may benefit the most from therapies aimed at reducing or eliminating Αβ from the brain before irreversible neuronal or synaptic loss occurs (Sperling et al. 2011b).
Individuals fulfilling criteria for MCI are a heterogeneous group with a wide spectrum of underlying pathologies (Petersen 2000; Winblad et al. 2004). About 40–60 % of carefully characterised subjects with MCI will subsequently progress to meet criteria for AD over a 3–4-year period (Petersen et al. 1995, 1999; Petersen 2000). Αβ imaging has proven useful in identifying MCI individuals with and without AD pathology. Approximately 50–70 % of the subjects classified as MCI present with high cortical 11C-PiB retention (Price et al. 2005; Kemppainen et al. 2007; Pike et al. 2007; Forsberg et al. 2008; Lowe et al. 2009; Mormino et al. 2009). While most studies found that subjects classified as non-amnestic MCI show low Αβ levels in the brain (Pike et al. 2007; Lowe et al. 2009), other reports did not (Wolk et al. 2009). More specifically, subjects classified as non-amnestic single-domain MCI usually do not show Αβ deposition, consistent with a non-AD underlying pathological process (Pike et al. 2007; Villemagne et al. 2011b).
To date, the relationship between Αβ deposition and the development of clinical symptoms is not fully understood. Although Aβ burden as assessed by PET does not correlate with measures of memory impairment in established clinical AD, it does correlate with memory impairment in MCI and healthy older subjects (Pike et al. 2007; Villemagne et al. 2008b). In subjects with MCI, this Aβ-related impairment is thought to be mediated through hippocampal atrophy (Mormino et al. 2009) while modulated by cognitive reserve (Roe et al. 2008; Rentz et al. 2010) and glucose metabolism (Cohen et al. 2009), although at the AD stage there is no longer correlation between glucose metabolism, cognition and Αβ burden (Furst et al. 2012).
Longitudinal studies are helping clarify the relationship between Aβ burden and cognitive decline in healthy individuals and MCI subjects. High 11C-PiB cortical retention in non-demented elderly subjects is associated with a greater risk of cognitive decline (Villemagne et al. 2008b, 2011b; Resnick et al. 2010). For example, the evaluation of either the cognitive trajectories (Villemagne et al. 2008b; Resnick et al. 2010) or changes in cognition prospectively (Morris et al. 2009; Villemagne et al. 2011b; Doraiswamy et al. 2012) shows that individuals with substantial Αβ deposition are more likely to present with cognitive decline. Aggregating the results from recently reported longitudinal studies reveals that 65 % of MCI subjects with marked 11C-PiB retention progressed to AD over 2–3 years, showing that MCI subjects with high Aβ deposition in the brain are >10 times more likely to progress to AD than those with low Aβ deposition, highlighting the clinical relevance and potential impact in patient management of Aβ imaging (Forsberg et al. 2008; Okello et al. 2009b; Wolk et al. 2009; Villemagne et al. 2011b). The observations that 11C-PiB retention in non-demented individuals relates to episodic memory impairment, one of the earliest clinical symptoms of AD, and that extensive Aβ deposition is associated with a significant higher risk of cognitive decline in HC and with progression from MCI to AD emphasise the non-benign nature of Aβ deposition and support the hypothesis that Aβ deposition occurs well before the onset of symptoms, further suggesting that early disease-specific therapeutic intervention at the presymptomatic stage might be the most promising approach to either delay onset or halt disease progression (Sperling et al. 2011b).
The high prevalence of Αβ deposition in non-demented individuals raises questions in regard to Αβ toxicity and deposition. The evidence points to the fact that Αβ toxicity in the form of oligomers and Αβ deposition in the form of aggregates precede by many years the appearance of clinical symptoms (Price and Morris 1999). These neurotoxic processes eventually lead to synaptic and neuronal dysfunction, manifested as cognitive impairment, grey matter atrophy and glucose hypometabolism (Eckert et al. 2003; Suo et al. 2004; Leuner et al. 2007). Therefore, it is highly likely that 11C-PiB retention in non-demented individuals reflects preclinical AD (Mintun et al. 2006; Rowe et al. 2007; Aizenstein et al. 2008; Villemagne et al. 2008b). This may be associated or attributed to a different susceptibility/vulnerability to Αβ, at either a cellular (frontal neurons seems to be more resistant to Αβ than hippocampal neurons) (Roder et al. 2003; Resende et al. 2007), a regional (compensatory upregulation of choline acetyltransferase (ChAT) activity in the frontal cortex and hippocampi of MCI and early AD subjects) (DeKosky et al. 2002) as well as at an individual or personal level, either due to a particular cognitive reserve (Mortimer 1997; Stern 2002; Kemppainen et al. 2008; Roe et al. 2008), differences in Aβ conformation affecting toxicity and/or aggregation (Lockhart et al. 2005; Deshpande et al. 2006; Levine and Walker 2010a; Walker et al. 2008), or because an idiosyncratic threshold must be exceeded for synaptic failure and neuronal death to ensue (Suo et al. 2004). These factors would help explain why some older individuals with a significant Αβ burden are cognitively unimpaired, while others with lower Αβ burden and no genetic predisposing factors have already developed the full clinical AD phenotype. Ongoing longitudinal studies will help identify those subjects that despite substantial Aβ deposition do no develop the AD phenotype, thus allowing to isolate genetic or environmental factors that may convey resistance to Aβ (Sojkova and Resnick 2011).
The lack of a strong association between Aβ deposition and measures of cognition, synaptic activity and neurodegeneration in AD, in addition to the evidence of Αβ deposition in a high percentage of MCI and asymptomatic HC, suggests that Αβ is an early and necessary, though not sufficient, cause for cognitive decline in AD (Villemagne et al. 2008b; Rabinovici et al. 2010; Sojkova and Resnick 2011), indicating the involvement of other downstream mechanisms, likely triggered by Αβ such as NFT formation, synaptic failure and eventually neuronal loss. Also at play are factors as age, years of education, occupational level and brain volume among others that, under the umbrella of ‘cognitive reserve’, appear to have a modulatory role between cognition and Αβ deposition (Roe et al. 2008; Chetelat et al. 2010b; Tucker and Stern 2011). Perhaps specific in vivo tau imaging will help elucidate or bridge the gap between Aβ deposition and the persistent decline in cognitive function observed in AD (Fodero-Tavoletti et al. 2011; Villemagne et al. 2012a).
10.6 Relationship of Aβ Imaging with Other Biomarkers
Another rapidly growing area is the exploration of the potential association between biomarkers of Aβ deposition or neurodegeneration (Wahlund and Blennow 2003; Morris et al. 2005; Sunderland et al. 2005; de Leon et al. 2006; Thal et al. 2006; Blennow et al. 2007; Shaw et al. 2007; Clark et al. 2008; Jack et al. 2010; Albert et al. 2011; McKhann et al. 2011; Storandt et al. 2012) and Aβ burden as measured by PET (Jack et al. 2008a, 2010).
10.6.1 FDG
While the typical pattern of Aβ deposition in sporadic AD is highest in frontal, cingulate, precuneus, striatum, parietal and lateral temporal cortices (Klunk et al. 2004), in FDG studies, the typical ‘AD pattern’ is of temporoparietal and posterior cingulate hypometabolism with sparing of the basal ganglia, thalamus, cerebellum and primary sensorimotor cortex (Salmon et al. 1994; Devanand et al. 1997; Coleman 2005; Jagust et al. 2007). Due to its high sensitivity (>90 %) for detecting temporoparietal and posterior cingulate hypometabolism, FDG-PET has improved diagnostic and prognostic accuracy in patients with probable AD (Salmon et al. 1994; Kennedy et al. 1995; Small et al. 1995; Silverman et al. 2001, 2002). A similar pattern of hypometabolism has been reported in normal elderly ApoE ε4 carriers (Reiman et al. 1996), MCI (Chetelat et al. 2003, 2005; Mosconi et al. 2006a), asymptomatic subjects with mutations associated with familial AD (Kennedy et al. 1995; Rossor et al. 1996) and subjects with a strong family history of AD (Mosconi et al. 2006b). FDG hypometabolism is correlated with cognition (Landau et al. 2011; Furst et al. 2012) and is predictive of future cognitive decline (Drzezga et al. 2003, 2005; Mosconi et al. 2004).
Visual assessment of PET images by clinicians blinded to the clinical status has demonstrated that 11C-PiB was more accurate than FDG to distinguish AD from HC (Ng et al. 2007a) and slightly better than FDG in differentiating AD from FTLD (Rabinovici et al. 2011). Similarly, Aβ imaging outperforms FDG at identifying MCI subtypes (Lowe et al. 2009).
While some reports found no association between FDG and 11C-PiB in AD (Furst et al. 2012), others found an inverse correlation between them in temporal and parietal cortices (Cohen et al. 2009), but no correlation has been shown in the frontal lobe (Klunk et al. 2004; Edison et al. 2007). A possible explanation for this dissociation is that there is a compensatory mechanism such as upregulation of ChAT in the frontal lobe of MCI individuals and mild AD patients (DeKosky et al. 2002; Ikonomovic et al. 2007) that might delay the manifestation of synaptic dysfunction; therefore, no hypometabolism is observed on FDG in the frontal lobe. Glucose metabolism in the frontal lobe eventually decreases as the disease progresses, and the characteristic temporoparietal pattern of AD becomes less apparent (Mielke et al. 1992; Herholz 1995). Conversely to what is observed in the frontal area, in AD patients, ChAT is significantly decreased in the posterior cingulate gyrus (Ikonomovic et al. 2011), which is characterised by being one of the regions where marked and early Aβ deposition is associated with substantial hypometabolism (Drzezga et al. 2011; Yokokura et al. 2011).
10.6.2 CSF
Analysis of CSF allows simultaneous measurement of the concentration of the two main hallmarks of AD, Aβ and tau. It has been reported to be highly accurate in the diagnosis of AD as well of predicting cognitive decline (Blennow et al. 2007; de Leon et al. 2007; Hansson et al. 2007; Mattsson et al. 2009, 2011). The typical CSF profile in AD is low Aβ1–42 and high total (t-tau) and phosphorylated tau (p-tau) (Strozyk et al. 2003; Hampel et al. 2010). Slower Aβ clearance from the brain (Bateman et al. 2006; Mawuenyega et al. 2010) is likely to lead to Aβ deposition in the brain and lower CSF Aβ1−42 concentrations. While there is no correlation between 11C-PiB retention and CSF t-tau or p-tau (Tolboom et al. 2009a; Forsberg et al. 2010), several studies have reported a strong inverse correlation between Aβ deposition in the brain as measured by 11C-PiB and CSF Aβ1–42 (Fagan et al. 2006, 2007, 2009; Forsberg et al. 2008; Koivunen et al. 2008a; Grimmer et al. 2009). Both high 11C-PiB retention and low CSF Aβ1–42 have been observed in cognitively unimpaired individuals probably reflecting Aβ deposition years before the manifestation of the AD phenotype (Mintun et al. 2006; Fagan et al. 2007, 2009; Rowe et al. 2007; Aizenstein et al. 2008). Large population studies such as ADNI (Shaw et al. 2007; Apostolova et al. 2010) will further validate the value of Aβ imaging and CSF assessments as antecedent biomarkers of AD.
10.6.3 MRI
While Aβ imaging or CSF Aβ provides information in regard to Aβ pathology, CSF tau quantification, FDG and structural MRI provide information related to the neurodegenerative process (Jack et al. 2010). Hippocampal and cortical grey matter atrophy along with ventricular enlargement are typical MRI findings in AD and MCI (Frisoni et al. 2010; Drago et al. 2011). Furthermore, it has been shown that the rates of hippocampal atrophy might be predictive of conversion from MCI to AD (Jack et al. 2008b).
Aβ deposition is associated with regional cerebral atrophy as measured by MRI (Archer et al. 2006; Bourgeat et al. 2010; Chetelat et al. 2010a; Becker et al. 2011) and correlates with the rates of cerebral atrophy (Becker et al. 2011; Tosun et al. 2011). Moreover, the data suggest that the relationship between Aβ deposition and cortical atrophy is sequential, where Aβ deposition precedes synaptic dysfunction and neuronal loss (Chetelat et al. 2010a; Drzezga et al. 2011; Forster et al. 2012), then manifested as structural changes (Becker et al. 2011). While one report did not find a correlation between atrophy trajectories and 11C-PiB retention in cognitively unimpaired elderly individuals (Driscoll et al. 2011), another reported that those with high Aβ deposition showed a significantly higher rate of atrophy in the temporal and posterior cingulate cortices compared to those with low Aβ burden (Chetelat et al. 2012).
10.6.4 Neuroinflammation
Microglia are the primary resident immune surveillance cells in the brain and are thought to play a significant role in the pathogenesis of several neurodegenerative disorders including AD (Venneti et al. 2006). Activated microglia can be measured using radioligands for the translocator protein (TSPO), formerly known as the peripheral benzodiazepine receptor. The TSPO is upregulated on activated macrophages and microglia and has been established as a biomarker of neuroinflammation in the central nervous system. Most reports found that in AD there is no association between Aβ deposition and microglial activation measured by [11C]PK11195 (Edison et al. 2008a; Okello et al. 2009a; Wiley et al. 2009) despite being an association between [11C]PK11195 binding and both glucose hypometabolism and severity of dementia (Edison et al. 2008a; Yokokura et al. 2011). More recently, PET studies assessing the relationship between Aβ deposition and neuroinflammation have been proposed using the MAO-B inhibitor L-deprenyl as a marker for astrocytosis (Carter et al. 2012).
10.7 Relationship of Aβ Deposition with Genetic Risks and Predisposing Factors
Age is the strongest risk factor in sporadic AD, with the prevalence of the disease increasing exponentially with age. These risks are increased in the presence of the ApoE ε4 allele (Farrer et al. 1997). Both risk factors have been directly associated with Aβ burden as measured by PET (Reiman et al. 2009b; Morris et al. 2010; Rowe et al. 2010).
To date, ApoE ε4 is the most consistent genetic risk factor associated with sporadic AD, and its presence has been associated with an earlier age of onset and a dose-dependent—either one or two ApoE ε4 alleles—higher risk of developing AD (Farrer et al. 1997). Examination of ApoE ε4 allele status revealed that, independent of clinical classification, ε4 carriers present with significantly higher 11C-PiB retention than non-ε4 carriers, further emphasising the crucial role that ApoE plays in the metabolism of Αβ (Reiman et al. 2009b; Morris et al. 2010; Rowe et al. 2010). When specific clinical groups are examined, the results—from both autopsy and Αβ imaging studies—are not that clear cut. While some studies found significant differences in Αβ burden between AD ε4 carriers and non-ε4 carriers (Gomez-Isla et al. 1996; Drzezga et al. 2008; Grimmer et al. 2010), others did not (Berg et al. 1998; Klunk et al. 2004; Rowe et al. 2007; Rabinovici et al. 2010).
Both symptomatic and asymptomatic individual carriers of mutations within the APP or presenilin 1 or 2 genes associated with familial AD present with very high and early 11C-PiB retention in the caudate nuclei, retention that seems to be independent of mutation type or disease severity (Klunk et al. 2007; Koivunen et al. 2008b; Remes et al. 2008; Villemagne et al. 2009a). Larger multicenter studies are helping elucidate the value of several biomarkers in these autosomal mutation carriers, assessment that might lead to better targeted disease-specific therapeutic strategies (Bateman et al. 2012).
While a similar pattern of Aβ deposition as seen in AD has been reported in older adults with Down syndrome (Landt et al. 2011), a completely different pattern of 11C-PiB retention was observed in a mutation carrier of familial British dementia (Villemagne et al. 2010).
There are several other factors that have been associated with AD. Among these, the most relevant are vascular risk factors (VRF), such as diabetes, hypercholesterolemia, hypertension and hyperhomocysteinemia. The influence of these VRF on AD pathology is an area that, despite the long and intense scientific debate, has not been clearly established (Viswanathan et al. 2009). While some studies support a direct role of VRF in the development of AD (Qiu et al. 2010), others suggest that they may accelerate its clinical presentation (Riekse et al. 2004). Recent studies have used MRI white matter hyperintense lesions as surrogate markers of vascular disease in association with assessment of Aβ deposition and cognition (Kuczynski et al. 2008; Marchant et al. 2012).
As with AD, VRF are strongly age-dependent factors that share similar pathogenic pathways, such as oxidative stress or inflammation, so it is not easy to extricate one from the other in order to ascertain a causal, casual or co-morbid association between them (Rosendorff et al. 2007). On the other hand, in light of the absence of disease-modifying therapies for AD, the relevance of these risk factors rests in the fact that early intervention over them, especially at middle age, might have an impact on either the onset or progression of AD.
10.8 Aβ Imaging in Other Neurodegenerative Conditions
Aβ imaging in a wide spectrum of neurodegenerative conditions has yielded diverse results. Although lower than in AD, similar patterns of 11C-PiB retention are usually observed in DLB (Rowe et al. 2007; Gomperts et al. 2008; Maetzler et al. 2009). Cortical 11C-PiB retention is also higher in subjects diagnosed with CAA (Johnson et al. 2007), while there is usually no cortical 11C-PiB retention in patients with FTLD (Rabinovici et al. 2007; Rowe et al. 2007; Drzezga et al. 2008; Engler et al. 2008). Aβ imaging has also facilitated differential diagnosis in cases of patients with atypical presentations of dementia (Ng et al. 2007b; Wolk et al. 2012a).
10.8.1 Cerebral Amyloid Angiopathy
CAA, characterised by Aβ deposits in and around the media of small arteries and arterioles of the cerebral cortex and leptomeninges, has been found to be present in most patients with AD (Jellinger and Attems 2005). To this date, neuropathological examination of the brain remains the only definitive method for diagnostic confirmation of CAA. However, the combination of Aβ imaging with 11C-PiB PET and T2* susceptibility-weighted (SWI) MR imaging (Knudsen et al. 2001) allows for the concomitant assessment of molecular and structural changes in vivo. The combination of cerebral microhaemorrhages (MH) and superficial siderosis (SS) has been suggested as radiological markers for CAA (Feldman et al. 2008). Aβ imaging studies have shown both processes may be intimately associated with Aβ deposition (Dierksen et al. 2010; Dhollander et al. 2011), preferentially in posterior areas of the brain, showing a distinct pattern of 11C-PiB retention than the one observed in sporadic AD (Johnson et al. 2007). Furthermore, focal Aβ deposition might be useful in predicting new MH in CAA patients (Gurol et al. 2012). While deep subcortical MH are generally associated with VRF, lobar MH (particularly posterior) (Pettersen et al. 2008) are usually attributed to CAA (Knudsen et al. 2001; Vernooij et al. 2008). Lobar MH are a frequent finding in AD patients or even in cognitively normal older individuals, and they are strongly associated with increasing age and Aβ deposition (Yates et al. 2011). This association between Aβ and vascular lesions has crucial implications not only for the selection and risk stratification of individuals undergoing anticoagulant therapies but also in those enrolled in anti-Aβ therapeutic trials (Weller et al. 2009).
Related posts:
 Impact of the IWG/Dubois Criteria for Alzheimer’s Disease in Imaging Studies
Impact of the IWG/Dubois Criteria for Alzheimer’s Disease in Imaging Studies
 PET and SPECT Studies of Ageing and Cardiovascular Risk Factors for Alzheimer’s Disease
PET and SPECT Studies of Ageing and Cardiovascular Risk Factors for Alzheimer’s Disease
 Single-Photon Emission Computed Tomography [Neuro-SPECT] Imaging of Brain Tumors
Single-Photon Emission Computed Tomography [Neuro-SPECT] Imaging of Brain Tumors
 PET Reveals Pathophysiology in Ischemic Stroke
PET Reveals Pathophysiology in Ischemic Stroke
 Linking Molecular Neurobiology to Therapeutic Approaches for Alzheimer’s Disease with PET
Linking Molecular Neurobiology to Therapeutic Approaches for Alzheimer’s Disease with PET
 Radionuclide Imaging Studies in Pediatric Neurology
Radionuclide Imaging Studies in Pediatric Neurology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


