Subtyptype and genetic abnormalities
Frequency (%)
Clinical implication
Estimated 5 years EFS (%)
B-lineage
Hyperdiploidy >50
20–30
Excellent prognosis with antimetabolite-based therapy
85–95
Hypodiploidy <44
1–2
Poor prognosis
30–40
Trisomies 4 and 10
20–25
Excellent prognosis with antimetabolite-based therapy
85–90
t(12;21)(p13;q22) ETV6-RUNX1 (formerly known as TEL–AML1)
15–25
Excellent prognosis with intensive asparaginase therapy
80–95
t(9;22)(q34;q11.2) BCR–ABL1
2–4
Imatinib plus intensive chemotherapy improve early treatment outcome
80–90 at 3 years
t(4;11) (q21;q23);MLL-AF4
1–2
Poor prognosis
t(1;19)(q23;p13)TCF3–PBX1
2–6
Increased incidence in blacks; excellent prognosis with high-dose methotrexate treatment; increased risk of CNS relapse in some studies
80–85
T-lineage
MLL-ENL
2–4
Favorable prognosis
80–90
Approximately 75 % of childhood ALL cases experience recurrent genetic abnormalities, including aneuploidy or structural chromosomal arrangements, detected by conventional karyotyping and fluorescence in situ hybridization (FISH), hence cytogenetic is important in diagnosis and as an indicator of response to therapy, thus playing a key role in risk stratification of patients for treatment [37–39].
In B-lineage ALL, the alterations including: hyperdiploidy (>50 chromosomes) seen in 25–30 % and has a good prognosis. Recurrent translocations including t(12;21) (p13;q22), encoding for ETV6–RUNX1 (TEL-AML1), which has a good prognosis, t(1;19) TCF3– PBX1 (E2A-PBX1), t(9;22) BCR–ABL1 (Philadelphia chromosome), and rearrangement of MLL at 11q23 to a diverse range of fusion partners [38, 40].
T-lineage ALL is characterized by activating mutations of NOTCH1 and rearrangements of transcription factors TLX1 (HOX11), TLX3 (HOX11L2), LYL1, TAL1, and MLL [41]. The frequency of NOTCH1 activating mutations in T-cell leukemia provides a compelling rationale for the use of either inhibitors of the Notch pathway. T-lineage ALL is induced by the transformation of T-cell progenitors and mainly occurs in children and adolescents. Although treatment outcome in patients with T-ALL has improved in recent years, a significant number of patients remain at a high risk of relapse, and few individuals survive when relapse [42] (Fig. 43.1).
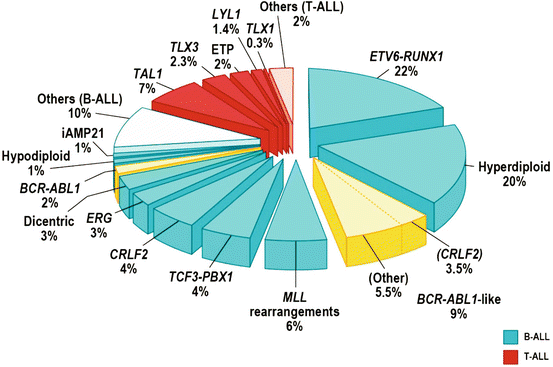
There are different subtypes of ALL based on morphological, immunological, cytogenetic, biochemical, and molecular genetic factors, and it may impact of the risk stratifications which have various responses to treatment.
43.3 Clinical Manifestation
Symptoms and clinical manifestations reflect of bone marrow infiltration by leukemic cells. Pallor, fever, muscle pain, bone pain, fatigue, bleeding (i.e. gum bleeding, epistaxis, ptechie, purpura, hematemesis and melena). The length of symptoms could be in weeks and occasionally several months. In physical examination organomegaly can be found [3, 44].
In complete blood count examination: Anemia, bicytopenia and often pancytopenia may be found.
Anemia: reflects of pressed erythropoiesis by young/immature leucocyte. Fever reflects infections due to low immunological status as peripheral blood dominated with young white blood cells (WBC), while WBC could be low, normal or high. Platelets: The platelets count usually low, and spontaneous bleeding can appear with platelet count 20,000–30,000/dL [3, 28, 45]. Summarized of clinical manifestations are listed in the Table 43.2.
Table 43.2
Clinical presentation of ALL
The clinical presentation of ALL: | |
|---|---|
Sign of anemia | Lethargy, weariness, fatigue, rapid exhaustion, lack of appetite. Laboratory: normocytic, normochromic anemia |
Signs of infections | Febrile illness. Laboratory: reduced of absolute neutrophil count |
Signs of bleeding tendency | Purpura, mucosal bleeding, hematomas and bruising. Laboratory: thrombocytopenia, occasional coagulopathy |
Signs of organ infiltration | Bone and join discomfort, hepatomegaly, generalized lymph node swelling, mediastinal mass and subsequent superior vena cava obstruction |
Signs of systemic disease | Fever of unknown origin, weight loss, night sweats |
43.3.1 Clinical Manifestations/Involvement in Other Systems
43.3.1.1 Central Nervous System Manifestations
Involvement of central nervous system (CNS) leukemia is defined by the presence of lymphoblast in the cerebrospinal fluid. It is found in 1.2–10 % of children with newly diagnosed ALL. Leukemic blasts entering the CNS by hematogenous spread. CNS leukemia is more common in mature B cell, T-ALL and children with high WBC. Signs of CNS involvement:
Signs of increased intracranial pressure (headache, papilledema and lethargy)
Signs and symptoms of parenchymal involvement (e.g., focal neurologic signs such as hemiparesis, cranial nerve palsies, convulsions, cerebellar involvement – ataxia, dysmetria, hypotonia, hyperflexia)
43.3.1.2 Cardiopulmonary Involvement
Leukemic involvement in the lungs and heart is rare. The manifestations could be: pericardial leukemic effusion and mediastinal mass, especially in T-ALL. Late cardiomyopathy is found after extensive treatment with anthracyclines [48].
43.3.1.3 Mediastinum
Mediastinal mass (thymus enlargement) due to leukemic infiltration, may cause life-threatening. Especially in T-ALL: superior vena cava syndrome.
43.3.1.4 Eye
Bleeding (retinal bleeding) caused by high white blood cell count and/or thrombocytopenia [28].
43.3.1.5 Musculoskeletal/Bone and Joint Manifestations
Involvement of musculoskeletal is characterized by severe pain, especially in lower extremities and sometimes unable/refusal to walk. This symptom occurred in 20–30 % of children with ALL. It may result of infiltration of leukemic cells to the bone or expansion of marrow cavity by leukemic cells. It may also appear swelling and tenderness due to leukemic infiltration [49, 50].
43.3.1.6 Urinary Tract (UT) Manifestations
Involvement of testicles is present mostly in boys. Testicular involvement is diagnosed if leukemic blasts found by testicular biopsy. It occurred only in boys with WBC >25,000/mm3, T-cell ALL, moderate to severe hepatosplenomegaly, lymphadenopathy and thrombocytopenia (<30,000/mm3). In girls, ovarian involvement occurs very rare [51–53].
43.3.1.7 Gastro Intestinal Tract (GIT) Involvement
The commonest manifestation of leukemia in GIT is bleeding, as reflected by occult blood in the stool. GIT bleeding might also be caused by thrombocytopenia, disseminated intravascular coagulation (DIC) or infection. Neutropenic typhlitis or necrotizing enterocolitis diagnosed if right lower quadrant pain with tenderness, abdominal tension, vomiting and sepsis are found [46].
43.3.2 Radiology Changes
Metaphysis: transverse radiolucent lines
Subperiosteal new bone formation
Osteolytic lesion involving medullary cavity and cortex
43.4 Diagnostic and Classification
Diagnostic of ALL is based on clinical findings and some laboratory tests. Basic investigation required for diagnostic ALL are [54–57]:
43.4.1 Blood Tests
Examination of complete blood count, differential blood count including morphology, lactate dehydrogenase (LDH), electrolytes, renal function tests, liver function tests, coagulation screening are necessary. Abnormal liver function test may be due to leukemic infiltration to the liver. Serum chemistry: Uric acid, potassium and calcium may be abnormal due to cell lysis as an impact of high WBC and chemotherapy. Serum lactate dehydrongenase usually high, and it may be has a prognostic value [58–60]. Morphology of leukemic cells can be examined from peripheral blood smear and bone marrow smear, hence morphology of peripheral blood and BM smear is critical.
Red blood cell and Hemoglobin: Normocytic; normochromic red cell morphology. Low hemoglobin indicates longer duration of leukemia.
White blood cell (WBC) count can be low, normal, or increased.
Blood smear: lympholasts are detected in children with high WBC. Very few to none (in patients with leukopenia). When WBC is greater than 10,000/mm3, blasts are usually abundant. Eosinophilia is occasionally seen in children with ALL; 20 % of patients with AML have an increased number of basophils.
Platelet. Thrombocytopenia: 92 % of patients have platelet counts below normal. Serious hemorrhage (Gastro Intestinal tract or intracranial) occurs at platelet counts less than 25,000/mm3.
43.4.2 Bone Marrow Tests
Diagnostic tools may vary among countries. It depends on the ability of each country to provide. The Important thing is morphological examination both from peripheral blood and bone marrow. Bone-marrow aspiration done under sterile conditions is recommended for diagnosis of acute lymphoblastic leukemia because morphology of leukemic cells in bone marrow can be different from those in peripheral smear and 20 % of patients with acute lymphoblastic leukemia do not have circulating blast cells at diagnosis [61]. Site of aspiration is recommended in the posterior iliac region for children above 2 years of age, and for children under 2 years at tibia. Sternal aspiration is contraindicated in young children.
43.4.2.1 Morphology
Leukemia must be suspected when the bone marrow contains more than 5 % blasts. The hallmark of the diagnosis of acute leukemia is the blast cell (more than 25 %), a relatively undifferentiated cell with diffusely distributed nuclear chromatin, one or more nucleoli, and basophilic cytoplasm. Special bone marrow studies, will help in detailed classification, include the following: Cytochemistry, Immunophenotyping, Cytogenetic and DNA content [55]. Bone marrow smear stained with other May-Grünwald-Giemsa or Wright-Giemsa, and should be examined under a light microscope. It is important to examine the morphology to distinguish lymphoblast and myeloblast. Acute leukemia can be classified based on morphologic characteristics into lymphoblastic leukemia and myeloblastic leukemia (Table 43.3a) [44].
Table 43.3a
Lymphoblast and myeloblast characteristic
Lymphoblast | Myeloblast | |
|---|---|---|
Cell size | 10–20 um | 14–20 um |
Cytoplasm | Blue, usually homogenous, sometimes with vacuoles | Blue-gray, granular, sometimes with Auer rods |
Nucleus chromatin | Homogenous and or fine | Heterogeneous |
Nucleoli | 0–2, distinct | 2–5 distinct “punched out” |
Nucleus/cytoplasm ratio | High | Low |
Cytochemical features is needed to sharpen the diagnosis [28]. Cytochemistry for myeloperoxidase and non-specific esterase should be done to exclude acute myeloid leukemia [62, 63]. To support diagnosis of ALL, cytochemistry such as periodic acid shift (PAS), peroxidase and Sudan-Black staining are recommended (Table 43.3b).
Table 43.3b
Cytochemistry characteristic of lymphoblast -myeloblast
Lymphoblast | Myeloblast | |
|---|---|---|
PAS | ++ | −/+ |
Sudan black | − | + |
Peroxidase | − | + |
Esterase | − | +/− |
43.4.2.2 FAB Classification
The French-American-British (FAB) Working Group Classification of ALL is based on morphologic and cytochemical features. Peripheral blood and bone marrow smears are stained (May-Grünwald-Giemsa method) and analyzed using light microscopy. Leukemic cells are characterized by a lack of differentiation, by a nucleus with diffuse chromatin structure, with one or more nucleoli, and by basophilic cytoplasm. This morphologic classification system categorizes lymphoblastic leukemias into three subtypes: L1, L2 or L3 (Table 43.4) [44].
Table 43.4
French-American-British (FAB) classification of lymphoblasts
L1 | L2 | L3 | |
|---|---|---|---|
Cell size | Small | Variable | Large, heterogeneous |
Nuclear shape | Regular, occasionally clefting | Irregular, clefting, indentation common | Regular, oval to round |
Chromatin | Homogenous | Variable, heterogeneous | Finely stippled and homogeneous |
Nucleoli | Not visible, | Often large, one or more present | Prominent, one or more |
Cytoplasm | Scanty | Variable, often moderately abundant | Moderately abundant |
Basophilic of cytoplasm | Very view | Variable, sometimes deep | Very deep |
Cytoplasmic vacuolization | Variable | Variable | Often prominent |
43.4.2.3 Immunophenotyping
Immunophenotyping of abnormal hematological cells using flow cytometry studies on peripheral blood or bone marrow samples improves both accuracy and reproducibility of classification of acute leukemias [64–69]. It is very useful for the diagnosis, classification, cost-effective treatment and prognostic evaluation in patients with hematological malignancies [64, 70–72]. Usually, ALL is classified into T-lineage, B-lineage, and B-cell (Burkitt’s) phenotypes. The World Health Organization (WHO) classification (Table43.5) divides ALL into two main groups only, i.e., B-lineage and T-lineage ALL, without further categorization [73–76].
Table 43.5
WHO classification of acute leukemia
Acute leukemias of ambiguous lineage |
Acute undifferentiated leukemia |
Mixed phenotype acute leukemia with t(9;22)(q34;q11.2); BCR-ABL1 |
Mixed phenotype acute leukemia with t(v;11q23); MLL rearranged |
Mixed phenotype acute leukemia, B-myeloid, NOS |
Mixed phenotype acute leukemia, T-myeloid, NOS |
Provisional entity: natural killer (NK) cell lymphoblastic leukemia/lymphoma |
B lymphoblastic leukemia/lymphoma |
B lymphoblastic leukemia/lymphoma, NOS |
B lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities |
B lymphoblastic leukemia/lymphoma with t(9;22)(q34;q11.2); BCR-ABL 1 |
B lymphoblastic leukemia/lymphoma with t(v;11q23);MLL rearranged |
B lymphoblastic leukemia/lymphoma with t(12;21)(p13;q22) TEL-AML1 (ETV6-RUNX1) |
B lymphoblastic leukemia/lymphoma with hyperdiploidy |
B lymphoblastic leukemia/lymphoma with hypodiploidy |
B lymphoblastic leukemia/lymphoma with t(5;14)(q31;q32) IL3-IGH |
B lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3); TCF3-PBX1 |
T lymphoblastic leukemia/lymphoma |
Classification of acute leukemia (B- or T-lineage ALL/AML) is based on reactivity patterns obtained with a panel of lineage-associated antibodies [77–79]. Immunophenotyping is also essential for distinguishing between ALL and AML; errors in differentiating between these two types of acute leukemias can occur in up to 10 % of cases [80–83]. Essential monoclonal antibodies for detecting acute leukemia are presented in Table 43.6.
Table 43.6
Monoclonal antibody panel for acute leukemia
B-linage | T-lineage | AML | |
|---|---|---|---|
Monoclonal antibody | CD10 | CD2 | CD13 |
CD19 | Cytoplasmic CD3 | CD33 | |
CD20 | CD5 | CD117 | |
CD22 | CD7 | Cytoplasmic MPO | |
CD34 | |||
Cytoplasmic CD79a | |||
HLA-DR | |||
IgM | |||
TdT |
The B-lineage phenotype ALL, positive for the following: B cell markers CD19, CD20, CD22, TdT, cytoplasmic CD79a, CD34 and CD10. It has been sub-classified according to maturation stage into: early pre B (pro-B), pre-B, transitional (or late) pre-B and (mature) B-ALL [64, 84]. In different regions, various incidences of B-lineage ALL have been reported. Burkitt’s leukemia displays an immunophenotype consisting of mature B cells [78].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


