Fig. 3.1
An overview of the mammalian cell cycle. A simplified figure of the cell cycle. Cyclin and cyclindependent kinases (CDK) are indicated, showing the various protein complexes generated during the cell cycle, their approximate abundances throughout the cell cycle are also indicated. Cyclin-dependent kinase inhibitors are indicated highlighting the specific cyclin/CDK complex that is inhibited. Each phase of the cell cycle is also shown
3.3 DNA Damage and Repair
Our DNA is continuously assaulted by a number of sources, including endogenous sources such as cell metabolism intermediates, replication fork collapse during regular DNA replication and repair events as well as exogenous sources such as the environment (for example ionizing radiation (IR) or exposure to genotoxic compounds). In addition the programmed endonucleolytic cleavage of DNA to yield double strand breaks (DSBs) is a natural component of meiotic DNA metabolism and immunoglobulin V(D)J gene rearrangement. DSBs are widely regarded as the most dangerous form of DNA damage, as the incorrect repair of DSBs causes genomic instability in the form of gross chromosomal loss, amplification, or rearrangements that can lead to cancer. In healthy cells, the harmful effects of DNA DSBs are controlled by large, multi-component macromolecular protein complexes, beginning with the detection of DNA damage and inducing complex protein signalling cascades ensuring genomic integrity.
As a consequence of this diverse range of threats, the cell and specifically the cell cycle is armed with DNA damage checkpoints that can stop the cell cycle following DNA damage allowing repair to occur to ensure the faithful transmission of the cells genetic information. These cell cycle checkpoints make certain that the DNA is correctly copied before the instigation of mitosis while the spindle assembly checkpoint inhibits anaphase until all of the chromosomes have been precisely aligned prior to separation. Crucial components of these cellular checkpoints act both directly and indirectly on cell cycle regulators to instigate a cell cycle arrest response as a facet of the DNA damage response (DDR).
Mammalian cells have evolved three mechanisms for the repair of DSBs (summarised in Fig. 3.2): single-strand annealing (SSA), Homologous recombination (HR) and non-homologous end-joining (NHEJ) [2–4]. Single strand annealing (SSA) repairs DNA by initially processing the DNA ends to yield overhangs (inevitably leading to large DNA deletions thus is highly error prone) allowing for searching, annealing, and ligation of homologous patches of DNA [5]. The SSA pathway is unique in that it does not require a separate similar or identical molecule of DNA and thus only requires a single DNA duplex, and uses the repeat sequences within eukaryote DNA as the identical sequence (that are required for homologous recombination) to drive repair. As DNA around the double-strand break site is cut, the single-stranded 3′ overhangs that are generated are bound by the RPA protein preventing the 3′ overhangs from sticking to themselves. Following RPA binding, the Rad52 protein is recruited to each of the repeat sequences on either side of the DNA break aligning them. This alignment enables the two complementary repeat sequences to anneal. After annealing is complete, leftover non-homologous flaps of the 3′ overhangs are cut away by the Rad1/Rad10 nucleases that are directed to the flaps by the Saw1 and Slx4 proteins. At this stage DNA synthesis occurs to complete any remaining gaps and ligation restores the DNA duplex as two continuous strands. The DNA sequence between the repeats is always lost, as is one of the two repeats. Even though there is the significant loss of genetic material during this process, SSA does have a role in DNA repair as the human genome is rich in repeat elements, for example there are over 106 Alu repeats in the human genome alone [6].
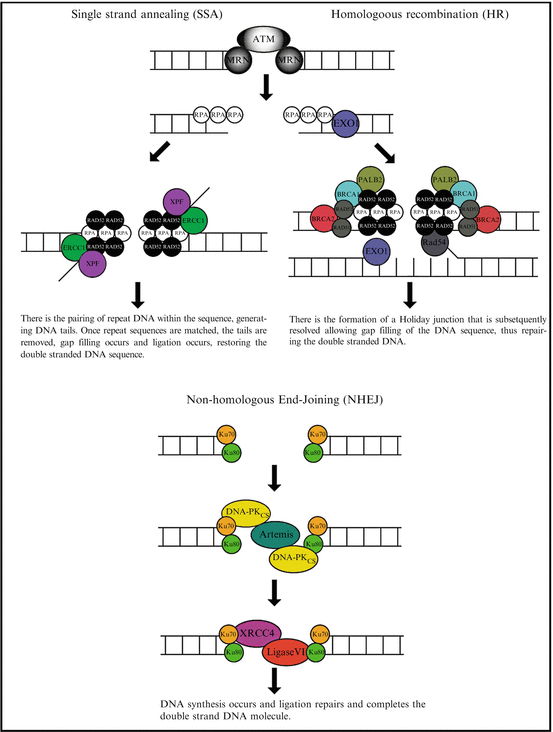
Fig. 3.2
An overview of the DNA repair process. The three fundamental repair processes in eukaryotic cells are highlighted. This includes single strand annealing, homologous recombination and non-homologous recombination
Homologous recombination (HR) is essential to cell division in eukaryotes and in addition to repairing DNA, HR also helps produce genetic diversity when cells divide during meiosis. Whether HR (or NHEJ) is used to repair double-strand breaks is largely determined by the phase of cell cycle. As HR requires an intact sister chromatid it is restricted to the S and G2 phases of the cell cycle [7]. After a DSB occurs, the MRN protein complex (consisting of Mre11, Rad50 and Nbs1) binds to the DNA on either side of the break after which a resection step occurs cutting back the DNA around the 5′ ends of the break. The MRN complex recruits the Ataxia-telangiectasia mutated (ATM) protein as well as the Sae2 protein to mediate signal transduction and generate these short 3′ overhangs of single-strand DNA. At this stage the 5′ to 3′ resection is continued by the Sgs1 helicase and the Exo1 nuclease. Once Sgs1 has opened the dsDNA sequence, the Exo1 nuclease function generates the ssDNA product. At this stage the RPA protein binds the 3′ overhangs. The PALB2, BRCA1, BCRA2, Rad51 and Rad54 proteins form a filament of nucleic acid and protein on the single strand of DNA coated with RPA. This nucleoprotein filament then begins searching for DNA sequences similar to that of the 3′ overhang. Once the matched sequence is found, the single-stranded nucleoprotein filament moves into (invades) the similar or identical recipient DNA duplex. A displacement loop (D-loop) is formed during this process and once it has occurred, DNA polymerase extends the end of the invading 3′ strand by synthesizing new DNA. This generates a Holliday junction. At this stage additional DNA synthesis occurs on the invading strand effectively restoring the strand on the homologous chromosome.
In contrast to SSA and HR, non-homologous end-joining (NHEJ) (which simply pieces together the broken DNA ends) is the predominant repair pathway in mammalian cells [7, 8]. This is because NHEJ does not require a complementary DNA sequence and therefore can be active during any stage of the cell cycle. In NHEJ repair, each broken DNA end is first bound by one Ku70/80 heterodimer, and two heterodimers must come together to bridge matching ends [9] ensuring high fidelity ligation. The resulting complex is subsequently bound by the DNA-dependent protein kinase catalytic subunit (DNA-PKCS), phosphorylating target proteins enabling NHEJ to proceed [10]. In vitro studies demonstrated that the Ku heterodimer initially binds to the DNA ends, translocate inwards in an ATP-independent manner and recruits DNA-PKCS stabilizing the protein/DNA binding [11–14]. Furthermore, DNA-PKCS can join two broken DNA ends together in a complex containing two DNA-PKCS molecules acting as a scaffold facilitating the re-joining [15, 16]. The remaining core of the NHEJ apparatus consists of the DNA ligase IV/XRCC4 (X-ray cross complementation group 4 protein) complex [17, 18]. The ligase IV/XRCC4 complex is essential for the ligation stage of NHEJ and is also thought to be involved in the alignment or gap filling of DNA prior to ligation [19]. XRCC4 has been shown to interact with DNA [20], Ku [21], DNA polymerase μ [22] and DNA-PKCS [18]. In addition to interacting with XRCC4, DNA-PKCS phosphorylates XRCC4 in vitro and in vivo [23, 24]. DNA ligase IV is an ATP-dependent DNA ligase with an amino-terminal catalytic domain that upon complex formation with XRCC4 stimulates its ligase activity [25].
However, these situations becomes significantly more complicated when one considers that regardless of source, DNA damage rarely produces clean breaks allowing straight forward blunt end ligation. Clearly the very nature of DNA damage ensures the cell is faced with a wide range of complex damage preventing efficient ligation presenting the requirement for further processing. The exposed 5′ and 3′ DNA ends are subject to resection and nucleotide addition/loss thus other components will be required for the NHEJ process to proceed efficiently. For example, the Werner syndrome protein (WRN) can remove 3′ phosphate or 3′ phosphoglycolate groups generated following IR and is itself phosphorylated by DNA-PK [26]. Interestingly, Artemis is a nuclease with 5′ to 3′ endonuclease activity that can remove 5′ overhangs and shorten 3′ overhangs [27] that is phosphorylated by DNA-PK activating the hair pin-opening activity of Artemis [28, 29]. Furthermore Ku80 has been shown to stimulates joining and artemis-mediated processing of DNA ends [30].
While NHEJ is a crucial process to repair DSBs generated by external sources, this process is also absolutely crucial for V(D)J recombination. This process is vital for antibody diversity and normal immune development and is the most widely investigated system for NHEJ (reviewed extensively in [31]). In combination with the RAG1/RAG2 proteins, DSBs are specifically generated. At these break sites, the Ku heterodimer binds to the free DNA ends of the DSB ensuring the spatial arrangement is preserved. DNA-PKCS binds the Ku/DNA complex, stimulating DNA-PK activity via phosphorylation enabling the NHEJ reaction to proceed. Furthermore the essential role of DNA-PK in DNA repair and preserving the genome is noted from the phenotype of defective/deleted cells. Cells that lack DNA-PKCS are acutely radiosensitive and have defective DSB repair (reviewed in [32]) while mice lacking DNA-PKCS remain viable although are immunodeficient (due to the absence of immune development) due to the accumulation of processed but not resolved DNA intermediates [33]. Furthermore DNA-PKCS–/– mice display significant telomeric fusion events consistent with DNA-PKCS role in telomere maintenance [34] (discussed below).
Just as it is imperative that our cells can detect and respond to DSBs, it is also crucial that our cells do not recognise the ends of our telomeres as dsDNA breaks. As such DNA-PK has been significantly implicated in telomere maintenance [35–38]. Mouse embryo fibroblasts obtained from DNA-PKCS–/– mice showed significant end-to-end chromosome fusion yet strikingly, these cells had sufficient telomere length and telomere DNA at the fusion sites [36, 38]. Following a number of yeast studies demonstrating a critical role of Ku at yeast telomeres [39, 40] it was demonstrated that Ku was present at the mammalian telomere [37, 41, 42]. The telomere/Ku complex is dependent upon the shelterin subunit TRF1, does not involve direct binding to TTAGGG telomeric repeat sequences [41, 43] and is independent of DNA-PKCS. Like Ku, DNA-PKCS is located at telomeres, has a role in telomere capping however does not affect either telomere length or telomerase activity, indicating that another function of DNA-PKCS is the protection of telomeric DNA and chromosome ends [34, 36, 38]. To date it is still unknown as to whether DNA-PKCS telomere recruitment is Ku-dependent and if DNA-PKCS role at the chromosome ends is structural. Furthermore the loss of DNA-PKCS has been shown to dramatically affect the rate of telomere loss in mice that lack both telomerase and DNA-PKCS compared to single knockout mice [44]. Additional studies revealed that this enhanced rate of telomere degradation was independent of Ku although the mechanistic relationship between DNA-PKCS and telomerase remains undefined [44].
As is clear, these cellular processes require a significant number of proteins and protein-protein complexes. Our cellular DSB repair pathways principally require ATM, the MRN protein complex, RPA, ATM- and Rad3-related (ATR), BRCA1, BRCA2 [45], Rad51, Rad52 Ku70/80, DNA-PKCS, Artemis and XRCC4.
3.4 The DNA Damage Response: Determining Cell Fate
In our cells the ability to repair DSBs is second only to the detection and response to DSBs. Within the cell DNA lesions are quickly recognized by the DNA damage response (DDR) proteins which activate cell cycle checkpoints and drive the repair process discussed previously. Depending on the nature and/or abundance of this damage different DNA repair pathways are involved, that together, form an extremely complex, interacting defense platform against genotoxic damage (summarized in Fig. 3.3). The DDR is a signal transduction pathway that is primarily mediated by proteins of the phosphatidylinositol 3-kinase-like protein kinases (PI3KKs) family many of which have been described in the repair of DSBs including, ATM, ATR and DNA-PK. In addition to these, there are also the poly(ADP) ribose polymerase (PARP) family. While there are 16 PARP family members, only PARP1 and PARP2 have been implicated in the DDR [46].
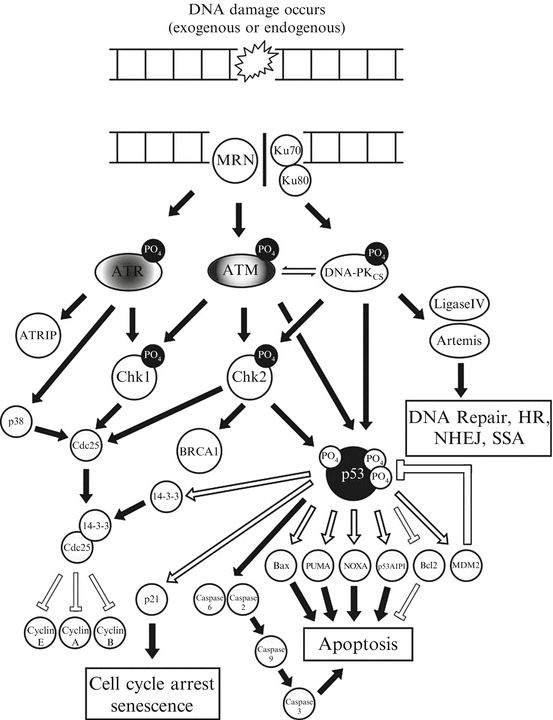
Fig. 3.3
A simplified schematic of the DNA-damage response. This figure indicates an overview of the key protein kinases and signalling intermediates within eukaryotic cells that are activated following the detection of a double strand break and the various cellular choices that they dictate, principally via the tumour suppressor and transcription factor p53
The DDR regulates all of the physiological processes that ultimately allow the cell to determine its fate; such as triggering apoptosis (programmed cell death), enter terminal differentiation via senescence (permanent cell cycle arrest) or to temporarily induce cell cycle arrest allowing DNA repair to occur. Taking into consideration the severity of these cellular choices a large proportion of the DDR is mediated by rapid post-translational protein modifications, such as phosphorylation or acetylation. While this is the case for the majority of the DDR signalling cascade, there is a proportion of this process that is mediated at the slower, transcription level, requiring various effecter gene transcription and subsequent protein translation prior to their involvement in the DDR. This dual action allows information to be incorporated within the DDR over time. Upon recognition of DNA lesions ATM, ATR and/or DNA-PK initially phosphorylate mediator proteins (including themselves) which act to amplify the DDR recruiting additional substrates including (but not exclusively) the Chk1, Chk2, p38 and MK2 kinases [47]. In addition to these, the most extensively studied component of the DDR is the tumour suppressor p53 which sits at the center of these signalling networks.
The transcription factor p53 is often referred to as “the guardian of the genome” as it is an essential regulator of the cellular response to stress and is crucial to the cellular DDR. Under normal physiological conditions the p53 protein is maintained at a low level by its negative regulator, the E3 ubiquitin ligase MDM2 that targets p53 for poly-ubiquitination and proteosomal degradation. However following the activation of the DDR, the p53-MDM2 interaction is disrupted and p53 is rapidly stabilized (following its initial phosphorylation at serine 15). The accumulated p53 protein can then undergo additional extensive post-translational modifications that includes further phosphorylation, acetylation, methylation, ubiquitination, sumoylation neddylation and glycosylation (reviewed extensively in [48]). Following DSB formation p53 is activated by ATM within a feedback loop that includes WIP1 phosphatase and MDM2, both of which are p53-regulated genes. This acts to turn off ATM and p53 respectively [49]. This temporal mechanism that activates p53-regualted gene expression in “waves” allows the cell to evaluate if the initiating damage has been repaired, suggesting that cell can obtain crucial cell fate information including the persistence of DNA damage, directing the cell to instigate apoptosis or senescence. This response is further enforced by the recognition of the DSB by the MRN complex, recruiting ATM and driving the HR process described previously. An important component of this process, highlighting the significant overlap within this cellular response is where DSB resection occurs after the RPA-DNA complex has formed. The recruitment of Rad51 to this complex, generates Rad51 filaments in a BCRA1-dependent manner driving HR. While this was considered to be exclusively ATM-dependent, Rad51 phosphorylation (by Chk1) is ATR-dependent [50] while BCRA2 itself is phosphorylated by ATR [51]. This indicates that both ATM and ATR are integral to the DNA repair and by their signalling to Chk1 and Chk2 potently activate p53, allowing p53 to dictate cell fate.
It is widely accepted that p53 activation triggers either cell cycle arrest or apoptosis and that it is the transcriptional activation of p53-regulated genes that is essential for tumour suppression. However, understanding how p53 can direct specific cell fates still remains elusive.
While the role(s) of DNA-PKCS in NHEJ and the DDR are clear, the most contentious issue regarding DNA-PKCS function involves DNA-PK signalling following cellular stress via the tumour suppressor protein p53. The waters become further muddied when one examines the considerable research focused on p53 and the vast cross-talk between different signalling cascades principally mediated by p53. While it is clear p53 can function in a transcription independent manner (for a review see [52, 53]) the clearest understandings of p53 function are based around its transcriptional activity [54]. The fact that over half of all cancers contain specific p53 mutations [55], the attenuation of p53-mediated gene expression clearly indicates the importance of p53-dependent gene expression in tumour suppression. The crucial limitation to date is how p53 turns particular genes on or off and has been the focus of intensive research [56–61].
Both in vitro and in vivo investigations have produced conflicting results with respect to and the involvement of DNA-PKCS in the signalling cascade that links DNA damage detection to p53 activation. Following any type of DNA damage the cell is faced with the decision to induce cell cycle arrest or induce apoptosis . This is further complicated with the reports implicating a role of DNA-PK and Ku in cellular senescence and autophagy [62, 63]. The stabilization and activation (via post-translational modifications) of p53 is crucial for each of these cell fates. It is now widely accepted that DNA-PKCS phosphorylates Chk2 (at threonine 68) [64, 65] and p53 at two specific residues (serine 15 and serine 37) [66] and there has been recent evidence that DNA-PKCS phosphorylates p53 at serine 46 [67–69]. Despite this clear p53 activation the role of DNA-PKCS in p53 activation remained controversial particularly in regard to the p53-dependent induction of cell cycle arrest [70–75]. In vivo studies using DNA-PKCS–/– mice categorically resolved this issue demonstrating that when absent, DNA-PKCS–/– mice could still phosphorylate p53 at serine 18 (the murine equivalent of human serine 15) following gamma irradiation (IR) and that fibroblasts from the these treated animals would undergo cell cycle arrest [76]. Further, these same groups demonstrated that it was the related PI3KKs ATM and ATR that mediated this cellular response [76].
However, the ability to induce apoptosis following DNA damage is critical to prevent cancer development and to prevent aberrant DNA from being passed to daughter cells after cell division. While it is now clear that DNA-PKCS does not have a role in inducing cell cycle arrest (discussed above) there is now a significant body of data implicating DNA-PKCS in the apoptotic response to severe DNA damage. For example, following the over expression of protein kinase Cδ normal cells mediate a robust apoptotic response. In contrast, DNA-PKCS–/– cells are significantly more resistant to this method of apoptosis induction [77]. This observation is further supported by studies showing that IR induced apoptosis (a p53-dependent process) is significantly attenuated in DNA-PKCS–/– mouse thymocytes [78]. Similarly following IR exposure E1A transformed fibroblasts mediate a potent p53-dependent apoptotic response that in the absence of DNA-PKCS was significantly attenuated [75, 79]. Concomitant to this observation, these DNA-PKCS–/– fibroblasts show significantly reduced p53 induction and the absence of p53 serine 18 phosphorylation [75]. In addition to mediating post-translational modifications, this was the first article to report that DNA-PK and p53 could, under these specific apoptotic conditions form a protein-protein complex [79]. Since this report, this observation was also noted in human myeloid leukemia, pancreatic and colon cancer cell lines after gemcitabine, a novel deoxycytidine analogue and current cancer therapeutic [80, 81]. These results suggest that DNA-PK and p53 may form a sensor complex that could detect the disruption of DNA replication caused by nucleoside analogue incorporation and may subsequently signal for apoptosis. These observations in particular support a number of immunohistological studies that show following IR, that ATM, ATR, p53 binding protein (p53BP1) and histone 2 AX (H2AX) form distinct DNA damage foci at the sites of DNA damage in contrast to both p53 and DNA-PKcs that show a diffuse nuclear staining profile [82]. These studies suggest that a p53-dependent apoptotic response could be directed by DNA-PKCS. Interestingly it has recently been shown that the p53-dependent apoptotic program requires (in addition to serine 15) serine 46 phosphorylation [83] a novel putative DNA-PKCS target residue [69]. Strengthening the case further, DNA-PKCS was shown to phosphorylate H2AX [84], a hallmark of apoptosis induction (for a detailed review see [85]). This report demonstrated that DNA-PK remained active in late apoptotic cells and that when active DNA-PK is able to initiate an early step in the DDR. DNA-PKCS has also been shown to negatively regulates p21 expression by directly interacting with the p21 transcription machinery via p53, thus priming the cell to induce apoptosis following cellular stress [81]. Recently it has been reported that the mechanism of killing during HIV viral integration is DNA-PK-dependent and activated (via phosphorylation) p53 and histone H2AX [86, 87]. Another study demonstrated that under cellular conditions that induced apoptosis, the inhibition of DNA-PKCS prevented p53 phosphorylation and accumulation, significantly reduced caspase-3 cleavage and attenuated the overall cellular apoptotic program [68]. Furthermore Ku70 was shown to accumulate after IR treatment and bound XIP8 correlating with reduced cell growth and elevated cell death [88]. The link between Ku70 and cell death is also noted in a neurodegenerative disease models where DNA-PKcs links DNA damage to Bax-dependent excitotoxic cell death, by phosphorylating Ku70 on serines 6 and/or 51, initiating Bax translocation to the mitochondria and directly activating a pro-apoptotic Bax-dependent death cascade [89]. These reports complement the described role of DNA-PK particularly in regard to the maintenance of chromosomes. As previously considered, telomerase deficient (Terc−/−) mice show widespread germ cell line apoptosis however a Terc–/–DNA-PKCS–/– double knockout mouse strain does not show increased apoptosis indicating a clear role in mediating apoptosis (that is independent of Ku) in cell lines with critically shortened telomeres [44, 90, 91].
3.5 The Clinical Significance
The loss of genomic integrity due to the loss or inactivation of DDR genes enhances the risk that cells will accumulate additional mutations that promote cancer development. This is strongly supported in data from several cancer types where the somatic mutations in DDR are routinely observed (summarised in Table 3.1). This is a significant component of many cancers in particular breast cancer where germline mutations in the DSB repair genes BRCA1 and BRCA2 significantly predispose carriers to developing breast and ovarian cancers. Similarly mutations in TP53 (a core component of the DDR) significantly predispose carries to childhood osteosarcoma, breast, brain, leukaemia and adrenocortical carcinomas. In addition to significantly increasing the predisposition to various cancers, mutations within the DDR also dramatically affect the sensitivity of tumours to chemotherapy. This has been most robustly demonstrated in HR and DSB repair deficiency where BRCA-deficient tumours are extremely sensitive to PARP inhibition. Clearly this is a double edged sword, while HR deficiencies could be effectively targeted by DSB-inducing chemotherapeutics, the genomic instability that enables the acquirement of additional mutations that could increase therapy resistance further. When treating cancer, the most significant aspect associated with chemotherapy are side-effects resulting from non-specific targeting to normal non-cancerous cell and poor efficacy as a result of intrinsic (such as mutated p53) or acquired drug resistance, such as a cellular change affecting drug metabolism or uptake. These aspects are considered in more detail in chapter W.LINK.
Table 3.1 Summary of the inherited genetic mutations within the DNA repair and cell cycle genes and the associated clinical syndrome. This table includes the most extensively studied genetic mutations that present as childhood cancers or significantly predispose individual’s to cancer
Syndrome | Gene(s) mutated | Clinical presentation | Mode of inheritance |
|---|---|---|---|
Fanconi anemia aplastic anemia, Myelodysplastic syndrome, Acute myeloid leukemia | FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN, FANCO, FANCP and BRCA2 | Hepatic tumors andsquamous cell carcinomas of the esophagus, oropharynx and uvula commonly present | Autosomal dominant |
Familial adenomatous polyposis | APC | Colorectal cancer | Autosomal dominant |
Hereditary breast-ovarian cancer syndrome | BRCA1, BRCA2 | Breast and ovarian cancer | Autosomal dominant |
Hereditary non-polyposis colon cancer (Lynch syndrome) | MLH1, MSH2, MSH6 and PMS2 | Colorectal, endometrial cancer, stomach cancer, ovarian cancer, cancers of the small bowel and pancreatic cancer | Autosomal dominant |
Hereditary paraganglioma-pheochromocytoma syndrome | Succinate dehydrogenase subunit genes, SDHD, SDHAF2, SDHC, SDHB | Neuroendocrine tumours | Autosomal dominant |
Li-Fraumeni syndrome | TP53 | Soft tissue sarcomas, osteosarcoma,breast cancer, brain cancer, leukaemia and adrenocortical carcinoma | Autosomal dominant |
MUTYH (mutY Homolog (E. coli))-associated polyposis | MUTYH |





