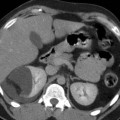
Fig. 1
Elongated right adrenal gland in a child with absent right kidney (arrows)
2.2 Horseshoe Kidneys
The horseshoe kidney is the most common fusion anomaly and its incidence is approximately 1:400 (Glenn 1959). The mechanism of fusion is unknown but it is thought to occur early in development after the ureteral bud enters the metanephric blastema. The kidneys lie on either side of the midline and their lower poles are connected by a band of either renal parenchymal or fibrous tissue (Fig. 2). This isthmus usually lies anterior to the great vessels. In a very small number of cases, the isthmus connects the upper poles, and rarer still is the “doughnut” kidney where both the upper and lower poles are fused.
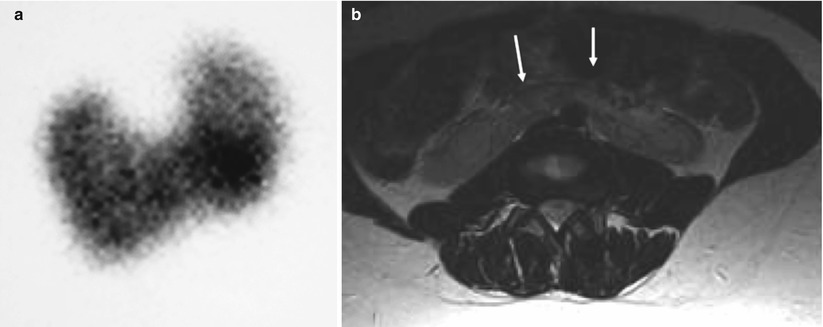
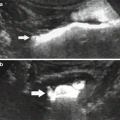
Fig. 2
(a) DMSA renogram demonstrating horseshoe kidneys with a connecting inferior band of renal tissue. (b) Connecting band in horseshoe kidneys identified incidentally in a patient who had an MRI of the spine (arrows)
The diagnosis can be made using renal ultrasonography. The lower poles of the kidneys are medially located and the renal pelvis may be anterior. Transverse images demonstrating the isthmus is essential to make the diagnosis and to distinguish from malrotation without fusion. Tc99m DMSA scan is excellent at identifying the orientations of the kidneys and the functioning renal tissue of the isthmus. It is especially useful when the amount of functioning renal parenchyma is small. If necessary the diagnosis can be confirmed using MRI, IVU or post-intravenous CT.
Thirty-three percent of patients with horseshoe kidneys have at least one other abnormality (Boatman et al. 1972). Associated anomalies include those of the central nervous system, gastrointestinal tract, cardiovascular and skeletal systems. Horseshoe kidneys have been reported in 20 % of patients with trisomy 18 (Boatman et al. 1972) and 7 % of patients with Turner’s syndrome (Lippe et al. 1988).
Hydronephrosis occurs in approximately 30 % of patients due to pelviureteric junction obstruction due to a high ureteral insertion or an anomalous renal vessel. Dilatation may also be due to reflux which has been noted in approximately 50 % of children with horseshoe kidneys. There is an increased incidence of Wilms’ tumour (Mesrobian et al. 1985).
2.3 Crossed Fused Ectopia
This is the second most common fusion anomaly after horseshoe kidney. The crossed ectopic kidney lies on the contralateral side to the insertion of its draining ureter into the bladder. Crossed ectopia with fusion occurs in 85 % of cases and without fusion in less than 10 % (McDonald and McClellan 1957; Abeshouse and Bhisitkul 1959).
Solitary and bilateral crossed renal ectopia are very rare. There are six types of crossed renal ectopia with fusion. The most common type is when the upper pole of the crossed kidney lies inferior to and is fused with the normally located kidney (Fig. 3). Its renal pelvis is anterior representing incomplete rotation. The next most common type is inferior ectopia where both pelves have opposite orientation indicating complete rotation.
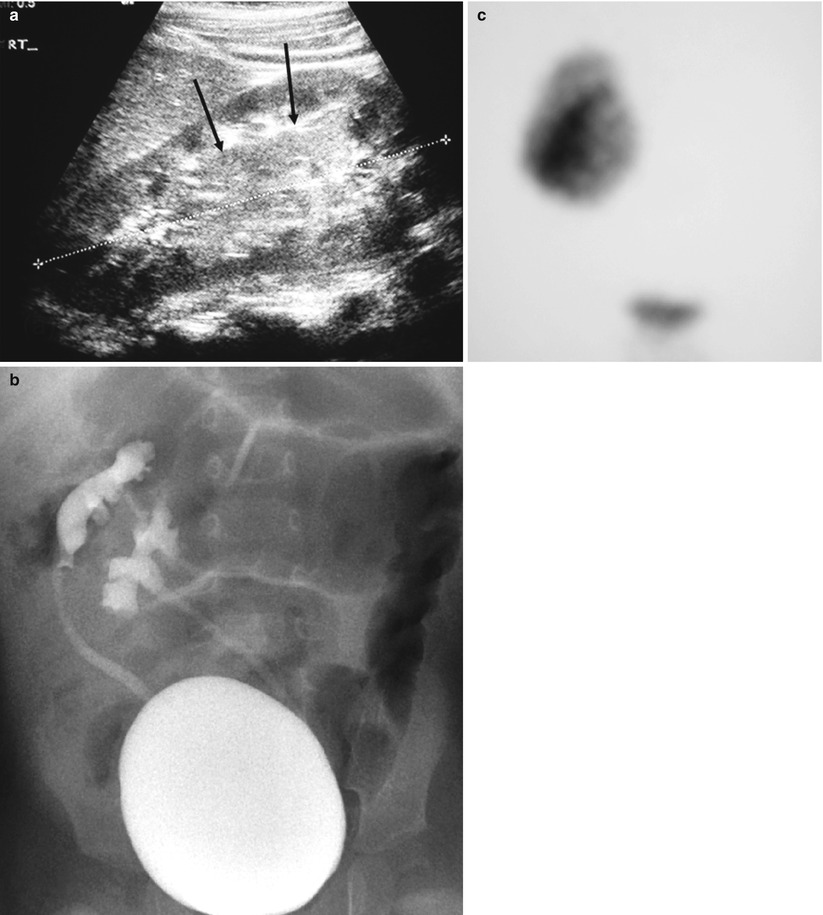
Fig. 3
Patient with crossed fused ectopia. (a) US at location of renal fusion (arrows). (b) VCUG demonstrating reflux into both collecting systems. (c) DMSA scan mimics a single right kidney
The diagnosis is suspected on sonography but it may be impossible to distinguish between fused and unfused kidneys. This may also be difficult to determine on intravenous urography because the poles often overlap. Tc99m DMSA scintigraphy with SPECT (Applegate et al. 1995), CT or MRI may be necessary to establish the diagnosis. MRI and CT angiography will also help to outline the vascular supply which may be quite variable. Vesicoureteric reflux is the most common associated anomaly and therefore a voiding cystourethrogram is indicated in these patients.
3 Renal Agenesis and Anomalies in Size
3.1 Renal Agenesis
Renal agenesis is the result of failure of the ureteric bud to connect with the metanephric blastema during the appropriate stage of development. A blind ending ipsilateral ureter of varying length may be present.
Bilateral renal agenesis is rare and estimated by Potter to occur in 1:48,000 cases (Potter 1972). The bladder is absent in approximately 50 % of cases, and when present the trigone is usually underdeveloped. The association of pulmonary hypoplasia, bowel anomalies, clubbed lower extremities and a typical facial appearance led Potter to describe the syndrome. There may also be associated genital anomalies.
Prenatally the diagnosis is usually made by sonography. There is severe oligohydramnios and failure to visualize foetal kidneys and a urinary bladder. The condition is incompatible with life.
Unilateral renal agenesis is often clinically silent. Ultrasonographic screening of 132,686 school children revealed an incidence of 1:12,800 children (Sheih et al. 1989).
The left kidney is absent more frequently than the right. There is aplasia of the ipsilateral adrenal gland in less than 10 % of cases. The contralateral kidney may be ectopic and malrotated or have vesicoureteric reflux. It may also be associated with genital abnormalities and is associated with syndromes such as Turners, Klippel–Feil, Polands and Mayer–Rokitansky.
When the diagnosis is made antenatally, postnatal sonography is required for confirmation and to evaluate the kidney on the opposite side. If the ipsilateral adrenal gland is present, it is often elongated (Fig. 1). There may be compensatory hypertrophy of the opposite kidney, but at birth the kidney may be normal in size and undergo compensatory hypertrophy later. Tc 99m DMSA scan is extremely useful in confirming the diagnosis and in assessing the size of the contralateral kidney. However, this study must be correlated with the ultrasound findings as a multicystic dysplastic kidney (MCDK) is also non-functioning and will mimic renal agenesis on scintigraphy.
As there is a strong association with vesicoureteric reflux, a voiding cystourethrogram is recommended in these infants. An MRI scan is useful in assessing associated genital anomalies (Lang et al. 1999).
3.2 Renal Hypoplasia
True renal hypoplasia, which is a small kidney with essentially normal renal parenchyma with fewer renal lobules and calyces and a normal ureter, is a rare congenital abnormality. It originates as a result of disturbed differentiation of metanephrogenic tissue or problems with the induction of tissue differentiation (Riccabona and Ring 2008). It may be combined with dysplastic elements. The kidney may be ectopic or malrotated and is prone to recurrent infections. Hypoplastic kidneys must be distinguished from acquired small kidneys such as those due to renal artery stenosis, renal vein thrombosis or infection.
Segmental renal hypoplasia or Ash-Upmark kidney is when one or more renal lobes fail to develop. Patients suffer from hypertension.
Renal hypoplasia may be diagnosed incidentally. Ultrasonography demonstrates small kidneys with a smooth outline (Fig. 4). In segmental hypoplasia the appearance may be similar to that of reflux nephropathy with indentation and thinning of the cortex together with calyceal dilatation and distortion. Some authors (Arant et al. 1979) believe that segmental hypoplasia is an acquired lesion as it is commonly associated with vesicoureteric reflux even in the absence of demonstrable infection.
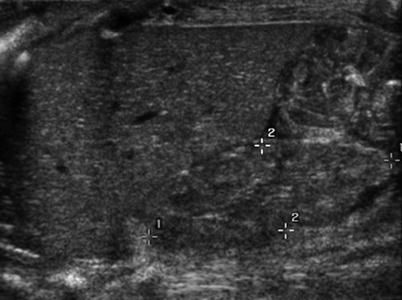
Fig. 4
Hypoplastic right kidney (between cursors). The kidney is small with a smooth outline
Voiding cystourethrography is recommended in patients with renal hypoplasia to out rule reflux. Tc 99m DMSA scintigraphy confirms the smooth outline of the kidney without focal defects. Intravenous urography is not indicated.
4 Renal Cystic Disease
Renal cystic disease can be divided into conditions of developmental origin such as MCDK and cystic renal dysplasia and conditions of genetic origin such as autosomal recessive polycystic kidney disease (ARPKD), autosomal dominant polycystic kidney disease (ADPKD), juvenile nephronophthisis (NPHP), medullary cystic kidney disease complex and glomerulocystic kidney disease (GCKD) (Avni et al. 2012)
4.1 Renal Cystic Disease of Developmental Origin
4.1.1 Multicystic Dysplastic Kidney
MCDK is the most common cystic renal lesion in children. A variety of proposed aetiologies have been associated with the underlying pathogenesis of MCDK. These include genetic disturbances, teratogens, in utero infections and urinary tract outflow obstruction (Hains et al. 2009; Lazebnik et al. 1999).
The incidence is 1:4,000 live births and is twice as common in males. The condition is usually unilateral and usually involves the entire kidney. It may be segmental (Jeon et al. 1999) or may involve one pole of a duplex system or one kidney in crossed ectopia (Kiddoo et al. 2005).
Abnormalities of the contralateral kidney are present in approximately one-third of patients (Lazebnik et al. 1999). These include vesicoureteric reflux (18 %) and pelviureteric junction obstruction (12 %) (Atiyeh et al. 1992). Other associated conditions may include ipsilateral genital anomalies and cardiac anomalies.
Nowadays most patients are detected antenatally by ultrasound screening. Foetal MRI may be helpful in difficult cases (Kiddoo et al. 2005) (Fig. 5).
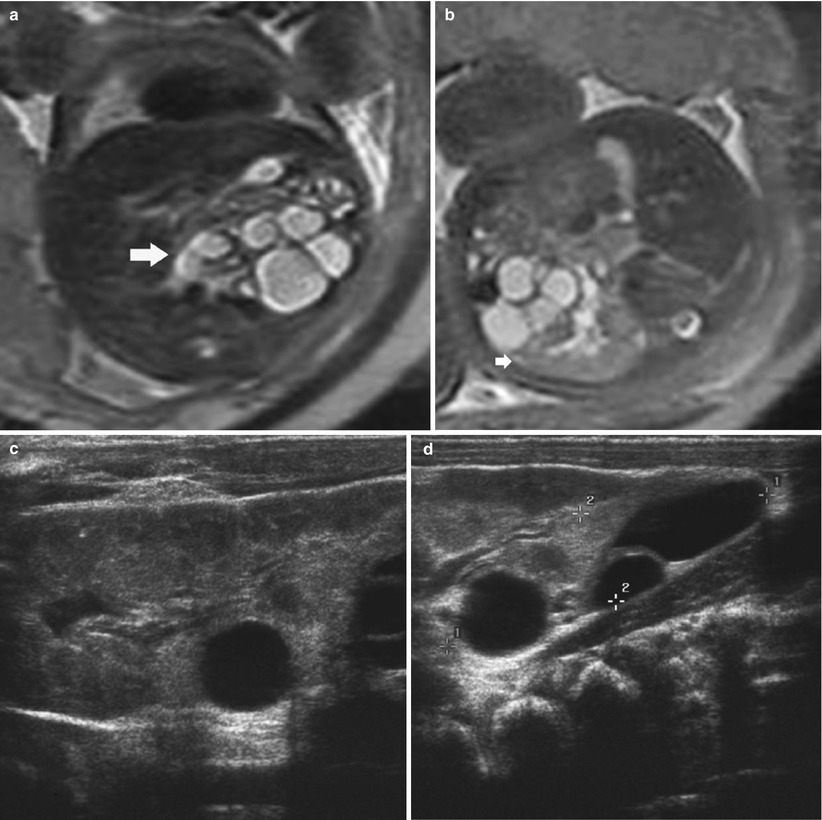
Fig. 5
T2 MRI in a foetus with crossed fused ectopia. (a) The anterior lower kidney demonstrates multiple cysts in keeping with a multicystic dysplastic kidney (MCDK) (arrow). (b) Normal posterior kidney (arrow). Postnatal US outlining the normal kidney (c) which is fused to the posterior MCDK (d)
Postnatal imaging is usually by ultrasonography to confirm the diagnosis and to assess the contralateral kidney. It shows a multicystic kidney with cysts of varying sizes which do not communicate, and the intervening renal tissue is echogenic (Fig. 6). Scintigraphy using Tc 99m DMSA shows a non-functioning kidney (Fig. 6). There is no general consensus on the routine use of voiding cystourethrography to determine if there is reflux into the contralateral kidney.
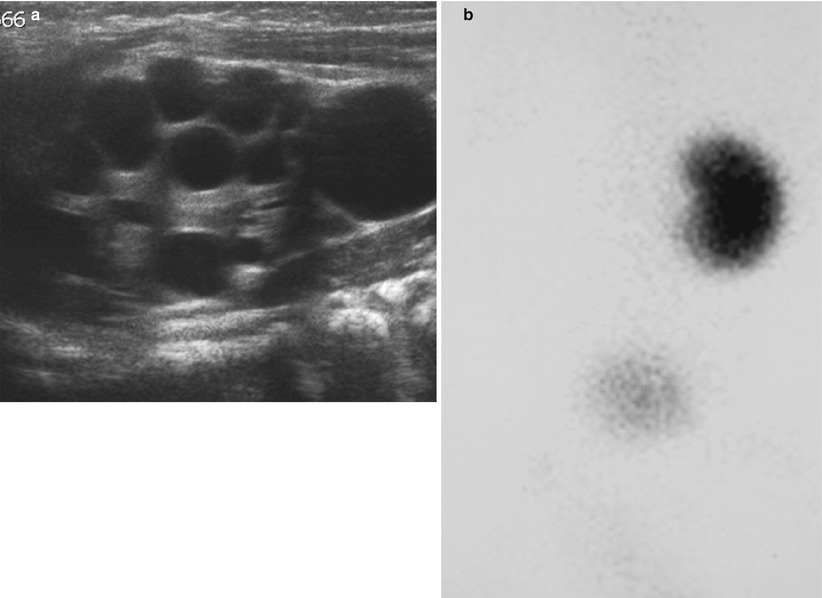
Fig. 6
Infant with MCDK. (a) US showing multiple cysts of varying size with intervening abnormally echogenic tissue. (b) DMSA scan confirms a non-functioning kidney on the same side
The management of MCDK is usually conservative as its natural history is that of spontaneous atrophy. Follow-up imaging is usually with ultrasonography. Wilms’ tumour has been reported in a patient with MCDK (Oddone et al. 1994), and hypertension has been reported in 23 % patients (Kiyah et al. 2009).
4.1.2 Cystic Renal Dysplasia
Renal dysplasia is the result of a developmental anomaly characterized by abnormal structural organization and development of metanephric elements. The kidneys may have multiple cysts. It is frequently associated with urinary tract obstruction and is seen in association with posterior urethral valves, prune belly syndrome and pelviureteric junction obstruction. This suggests that obstruction and urinary retention may cause abnormal renal development (Shibata and Nagata 2003).
Antenatal MRI may compliment antenatal ultrasonography in establishing the diagnosis (Fig. 7).
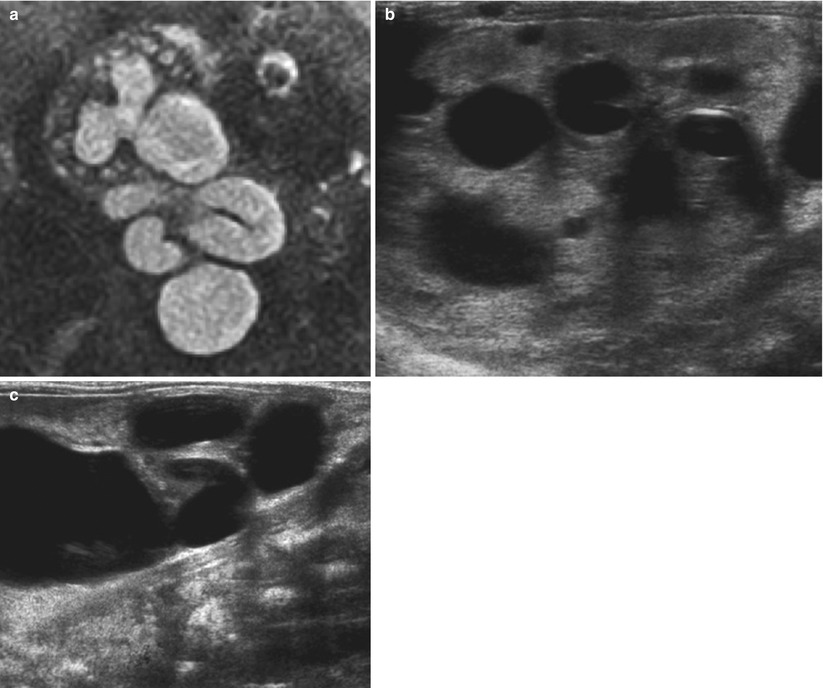
Fig. 7
(a) Foetal T2 MR showing a single kidney with significant hydronephrosis and hydroureter. There are multiple small cysts in keeping with cystic dysplasia (b, c). Postnatal US confirms the diagnosis
Postnatally on ultrasonography the kidneys are usually normal to small in size with increased cortical echogenicity, loss of corticomedullary differentiation and scattered small cysts (Fig. 8). Voiding cystourethrography may be indicated. Tc 99m MAG3 renography is helpful to confirm if there is associated obstruction to drainage, and Tc 99m DMSA may be required to assess functioning renal tissue.
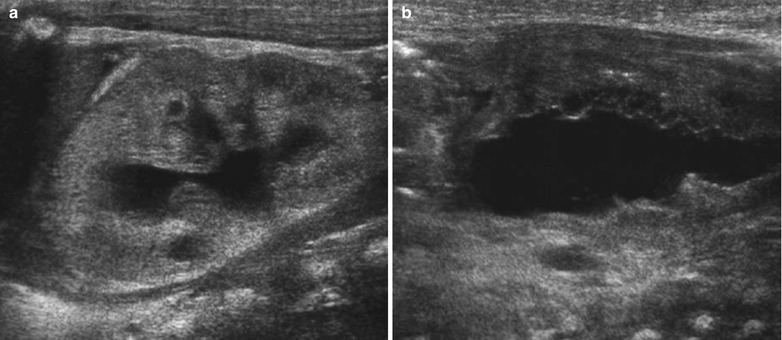
Fig. 8
Newborn with cystic renal dysplasia secondary to posterior urethral valves. (a) Left kidney: showing increased cortical echogenicity with loss of the corticomedullary differentiation and multiple cysts. There is renal collecting system dilatation. (b) Very irregular thick-walled urinary bladder due to posterior urethral valves
4.2 Renal Cystic Disease of Genetic Origin
4.2.1 Autosomal Recessive Polycystic Kidney Disease
ARPKD is an inherited disease characterized by nonobstructive fusiform dilatation of the renal collecting ducts resulting in enlarged kidneys. Liver disease is present in all patients with the manifestations varying according to the patient’s age at presentation. The principal pathologic features are periportal fibrosis and biliary duct ectasia. When there is significant liver involvement, it is referred to as congenital hepatic fibrosis (Turkbey et al. 2009). The condition is often referred to as infantile polycystic disease as over 90 % of patients suffer from renal insufficiency during childhood. It occurs in approximately 1:40,000 live births with a gene defect on chromosome 6 (Zerres et al. 1994).
Antenatally there may be oligohydramnios, very large kidneys and absence of bladder filling. MRI may compliment the ultrasound findings (Cassart et al. 2004).
Infants who are severely affected may have pulmonary hypoplasia and Potter’s phenotype and may die from respiratory failure. In selected cases removal of one or both kidneys may improve the respiratory status but the child will usually require dialysis.
Postnatal ultrasonography usually shows enlarged echogenic kidneys with no corticomedullary differentiation. Small cysts up to 3 mm in diameter may be seen and these are usually located peripherally (Boal and Teele 1980). The cysts may be too small to be seen and the echogenic pattern may have a speckled appearance (Fig. 9). As the child gets older, the kidneys may increase further in size and the cysts may enlarge. A family history helps to confirm the diagnosis. Further imaging is only required to follow the condition and this is best performed with sonography. Monitoring of hepatic involvement is necessary to detect complications of portal hypertension. The combined use of conventional and high-resolution ultrasonography with MR cholangiography allows detailed definition of the extent of the kidney and hepatobiliary manifestations without requiring ionizing radiation and contrast agents (Turkbey et al. 2009) (Fig. 9).
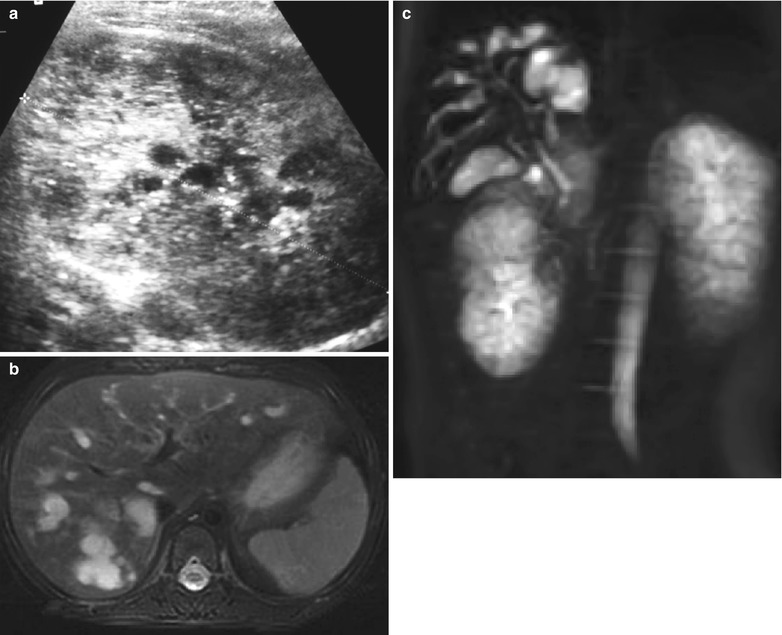
Fig. 9
Newborn with ARPKD. (a) The kidney is large, echogenic and contains multiple cysts, some of which are tiny giving rise to a speckled appearance. (b) Axial T2 MRI and (c) volume acquisition in the same patient at 2 years showing significant biliary ductal ectasia
4.2.2 Autosomal Dominant Polycystic Kidney Disease
ADPKD occurs in approximately 1:1,000 live births. The specific form which develops depends on which of the three genes, PKD1, PKD2 or PKD3, become mutated. In 90 % of patients, the affected gene is located on the short arm of chromosome 16.
The condition may be detected during infancy and childhood either by selective screening where there is a family history or incidentally. It usually presents during adulthood and is often referred to as adult polycystic kidney disease.
The cysts arise from the nephrons and collecting tubules with which they communicate directly, and islands of normal parenchyma are interspersed between the cysts.
Ultrasonography is the imaging modality of choice to investigate these patients. In infants and children, the kidneys may be normal but cysts can be seen in the neonatal period (Fig. 10). The cysts increase in number over time, may become very large and irregularly distributed throughout the kidney and these may compress normally functioning renal tissue. This will be seen as hyperechoic parenchyma on sonography. The cysts may be complicated by secondary haemorrhage, infection or rupture. Complimentary CT or MRI is rarely necessary.
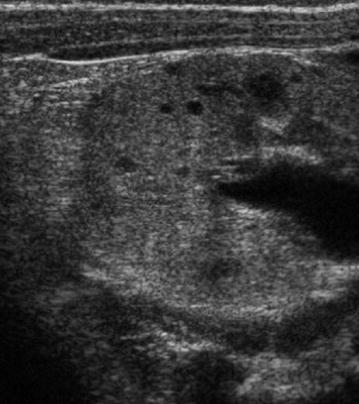
Fig. 10
Transverse US view of kidney in an infant with strong family history of ADPKD. There is some loss of the corticomedullary differentiation and multiple small cysts
Intracranial aneurysms associated with the condition have been described in neonates and young children (Proesmans et al. 1982). These may be diagnosed using MR angiography.
4.2.3 Medullary Cystic Disease and Nephronophthisis
Medullary cystic disease (MCDC) and NPHP are grouped together as they share many features. NPHP is an autosomal recessive disorder with early onset symptoms, and medullary cystic kidney disease is inherited as an autosomal dominant disorder which usually affects patients in their 30s.
Pathologically they cause cysts restricted to the renal medulla or corticomedullary junction in addition to tubular atrophy, tubular basement membrane disintegration and interstitial fibrosis.
Extrarenal manifestations of NPHP include oculomotor apraxia (Cogan syndrome), retinitis pigmentosa (Senior–Loken syndrome), cone-shaped epiphyses and liver fibrosis (Mainzer–Saldino syndrome), optic nerve coloboma with cerebellar vermis hypoplasia (Joubert syndrome type B) and cranioectodermal dysplasia and electroretinal abnormalities (Sensenbrenner syndrome).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


