Anti– N -methyl- d -aspartate (NMDA) receptor encephalitis is a severe but potentially reversible neurologic disorder that is clinically recognizable in children and adolescents. Prompt diagnosis and treatment are essential to facilitate recovery. Treatment consists of corticosteroids, intravenous immunoglobulin, or plasma exchange as first-line therapy followed by cyclophosphamide or rituximab, if necessary, as second-line immunotherapy. Patients with tumor-associated encephalitis benefit from tumor resection. More than 75% of patients make a substantial recovery, which occurs in the reverse order of symptom presentation associated with a decline in antibody titers.
Key points
- •
Anti– N -methyl- d -aspartate (NMDA) receptor encephalitis is a severe but potentially reversible neurologic disorder that is clinically recognizable in children and adolescents.
- •
Prompt diagnosis and treatment are essential to facilitate recovery.
- •
The clinical syndrome is confirmed by the presence of NMDA receptor antibodies in cerebrospinal fluid or serum.
- •
Treatment consists of corticosteroids, intravenous immunoglobulin, or plasma exchange as first-line therapy followed by cyclophosphamide or rituximab, if necessary, as second-line immunotherapy. Patients with tumor-associated encephalitis benefit from tumor resection.
- •
More than 75% of patients make a substantial recovery, which occurs in the reverse order of symptom presentation associated with a decline in antibody titers.
Introduction
In 2005, 4 young women with a syndrome of acute psychiatric symptoms, seizures, memory loss, encephalopathy, and hypoventilation associated with an ovarian teratoma were reported. These women were found to have antibodies that reacted with neuronal cell-surface antigens. These antibodies were subsequently found out to be autoantibodies targeting the NR1 and NR2 N -methyl- d -aspartate (NMDA) glutamate receptors in another series of 12 women with a similar neuropsychiatric syndrome in the presence of ovarian teratoma. These reports were just the beginning of what has now turned out to be an increasingly reported new immune-mediated neurologic syndrome, anti-NMDA receptor (NMDAR)-mediated encephalitis. A seminal publication in 2008 by Dalmau and colleagues described the syndrome in great detail and outlined its salient characteristics in a large cohort of 100 patients. The majority of reported subjects in the cohort were adult females who had an ovarian teratoma in the background. Since this article was published, however, it has become increasingly clear that the syndrome is not restricted to adult females but can also be seen in adult males and children of either sex in the absence of an associated systemic neoplasm. It appears that this syndrome is still underrecognized as its existence is only being gradually appreciated. However, other previously reported disorders could perhaps have been anti-NMDAR encephalitis; these include idiopathic encephalitis with psychiatric manifestations or dyskinesias, coma associated with intense bursts of abnormal movements and long-lasting cognitive and behavioral disturbances, acquired reversible autistic syndrome in acute encephalopathic illness in children, immune-mediated chorea encephalopathy syndrome in childhood, juvenile acute nonherpetic encephalitis, and encephalitis lethargica. Moreover, with increasing literature the syndrome appears to encompass a far wider spectrum of clinical presentation than initially described. The discovery of anti-NMDAR encephalitis has led to the detection of a growing list of many other autoimmune synaptic receptor encephalitides, including those mediated by antibodies toward the 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid (AMPA) receptor, γ-aminobutyric acid (GABA) B receptor, and leucine-rich, glioma-B inactivated 1 (LGI1) receptor. The LGI1 receptor is the antigen previously ascribed to voltage-gated potassium channel encephalitis. These immune-mediated responses are now providing an understanding of the function of the neurotransmitter receptors affected by these antibodies. This article focuses on anti-NMDAR encephalitis in the pediatric age group.
Epidemiology
The incidence of anti-NMDAR encephalitis in both children and adults is unknown at present. It appears to be more common than previously thought, given the increasing literature. A retrospective study of adults with encephalitis of unknown origin identified NMDAR antibodies in 1% of patients admitted to intensive care. A multicenter, population-based prospective study of causes of encephalitis in adults and children in the United Kingdom showed that 4% of patients had anti-NMDAR encephalitis. The most common immune-mediated causes were acute disseminated encephalomyelitis, followed by anti-NMDAR encephalitis and other antibody-associated encephalitides. The California Encephalitis project tested 20 patients and identified 10 as anti-NMDAR positive. All 10 patients had negative viral studies, but 4 were positive for serum Mycoplasma immunoglobulin M antibodies. The median age was 18.5 years, with a predilection for Asians and Pacific Islanders. In a case series in children and adults in 2009, 40% were children younger than 18 years with the youngest child being only 23 months old. In the Dalmau series of 100 patients, 22 were children younger than 18 years, 55% of whom had an underlying tumor. A child as young as 20 months has been reported with symptoms and neuronal antibodies consistent with anti-NMDAR encephalitis.
Clinical presentation
Anti-NMDAR encephalitis presents as a characteristic syndrome that develops in progressive stages of illness and recovery. The clinical syndrome was first defined in a series of 100 patients, which included adults and children. This study was followed by a second series that concentrated on 32 children younger than 18 years who were compared with 49 adults identified at the same time. Individual case reports and smaller case series have substantiated these findings. The phenotype in adults and children is similar, except that autonomic dysfunction and hypoventilation are probably less common and less severe in children.
Prodromal Features
At the onset of the syndrome, symptoms including fever, headache, rhinitis, vomiting, and diarrhea have been recorded in 48% of children.
Psychiatric Features
Within 2 weeks, patients usually develop psychiatric symptoms, including anxiety, paranoia, fear, psychosis, mania, and insomnia. Social withdrawal and stereotypical behavior are also possible. In a series of mostly adult patients, 85% presented initially to a psychiatrist for anxiety, agitation, and visual or auditory hallucinations. Of 32 children, 87% presented with changes in behavior or personality. The recognition of psychosis in young children is challenging. The behavioral changes include increased tantrums, irritability, and hyperactivity. Behavior can be hypersexual or aggressive. Acute mania with psychosis and a catatonic state has been reported in an adolescent female with anti-NMDAR encephalitis. As patients with psychosis are often treated initially with neuroleptic medications, neuroleptic malignant syndrome can compound the clinical picture because symptoms of rigidity, autonomic instability, and elevation of muscle enzymes, typically associated with this syndrome, can occur independently in anti-NMDAR encephalitis in the absence of neuroleptics.
Neurologic Features
In children, the chief features include movement disorders, seizures and cognitive problems. Other features reported in adults appear to be less common in children such as autonomic instability and sleep disorders.
Movement disorders
Movement disorders are a very frequent neurologic feature of this syndrome and have been reported in 84% of a case series in children with orofacial dyskinesias occurring in 45%, choreoathetosis in 32%, and dystonia in 32%. Oculogyric crisis, rigidity, and opisthotonic postures are also reported. Orofacial dyskinesias are described as chewing, tongue thrusting, lip smacking, and facial grimacing movements. There are descriptions of more complex, stereotyped movements including pelvic thrusting, pseudo–piano-playing motions, and writhing of the extremities. Some movements, such as pelvic thrusting, have the potential to be considered as nonorganic phenomena. Limb movements mimicking epileptic seizures may also occur. Patients may develop episodic opisthotonus, dystonia, and oculogyric crises, associated with tachycardia and hypertension, which is suggestive of autonomic storming. Opsoclonus-myoclonus has been reported in a woman with anti-NMDAR encephalitis.
Seizures
Seizures are reported in about 77% of children, and can be difficult to recognize and treat. Both partial and generalized seizures as well as status epilepticus can occur, although partial seizures seem to predominate. Abnormal repetitive movements may mimic partial seizures, necessitating video electroencephalogram (VEEG) monitoring for definitive diagnosis of seizures in some cases. The presentation of partial status epilepticus in anti-NMDAR encephalitis may lead to the consideration of a diagnosis of Rasmussen syndrome. Unexplained new-onset epilepsy in young women and adolescents may be a feature of anti-NMDAR encephalitis.
Cognitive problems
Short-term memory loss is underestimated, as the assessment of memory is affected by the psychiatric symptoms and speech problems. Retrospectively, patients often report amnesia of the illness. A rapid deterioration of language, ranging from reduced verbal output and echolalia with echopraxia to frank mutism, is common. This symptom may be followed by a decreased level of consciousness and alternate periods of agitation and catatonia. Dissociative responses to stimuli are often noted.
Autonomic Dysfunction
Tachycardia, hypertension, and hyperthermia occurred in 86% of a case series of 32 children. Autonomic instability and hypoventilation are reported to be less severe, and dysrhythmias in children are less likely to require pacing than in adults. Mechanical ventilation may sometimes be needed for central hypoventilation and airway protection. Hyperthermia is common throughout the disease, frequently leading to extensive investigations to exclude infectious processes. Hypersalivation and urinary incontinence are some of the other frequently reported symptoms.
Miscellaneous Features
Insomnia is reported as an early presenting sign, at least in adults. Sleep-wake cycles are disrupted and patients arouse frequently. During recovery patients may have hypersomnia. Symptoms of hypothalamic dysfunction have been recognized in a few patients. Recurrent optic neuritis with transient cerebral lesions has been reported in one patient.
Anti-NMDAR encephalitis has also been reported in an adolescent female, with recurrent relapses associated with extensive longitudinal myelitis and optic neuritis mimicking neuromyelitis optica.
Physical Examination
Systemic and neurologic signs are nonspecific. There are no markers in the clinical examination that can suggest anti-NMDAR encephalitis. Hence, a low threshold of suspicion is warranted in patients with a constellation of movement disorder, seizures, and neuropsychiatric problems. Patients show signs of a diffuse encephalopathy, indicating that there is neurologic dysfunction of subcortical, limbic, and frontostriatal circuitry. Various levels of altered consciousness are possible. Signs of increased intracranial pressure are usually uncommon, although they may be secondarily present in the wake of prolonged status epilepticus. Neurologic examination may reveal nonspecific signs of diffuse cerebral dysfunction such as exaggerated deep tendon reflexes, extensor plantar responses, and tone abnormalities, in addition to movement disorders. Soft neurologic signs such mild ataxia and difficulties with fine motor coordination can also be seen.
Investigations
Neuroimaging
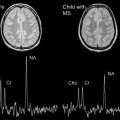
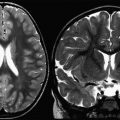
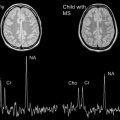
Computed tomography (CT) of the head is not useful because of its poor sensitivity. Magnetic resonance (MR) imaging of the brain is unremarkable in 50% of cases. In the other 50%, MR imaging may show nonspecific T2 or fluid-attenuated inversion recovery (FLAIR) signal hyperintensity within the hippocampus; cerebellar, frontobasal, or insular cortex; basal ganglia; brainstem; and, occasionally, the spinal cord. These changes are usually mild or transient, and have been associated with contrast enhancement in the hyperintense areas or the meninges. Occasionally the MR imaging shows extensive T2 FLAIR abnormalities. The radiologic differential diagnosis with these abnormalities remains broad. Follow-up brain MR imaging either remains normal or shows minimal change. Patients with refractory seizures have been reported to show brain atrophy. Lesions can be transient and nonenhancing, and may have a demyelinating appearance. MR spectroscopy has not yet been documented to be of any proven value. In short, no clear patterns of brain involvement are evident in this condition ( Figs. 1–3 ), which makes a diagnosis of anti-NMDAR encephalitis based solely on neuroimaging features rather difficult.



Functional Neuroimaging
Most of the literature on functional neuroimaging in anti-NMDAR encephalitis is case based and anecdotal. The utility of functional neuroimaging modalities has not been systematically studied. 18 F-Fluorodeoxyglucose positron emission tomography (FDG-PET) imaging showed cortical hypometabolism in 2 young girls aged 3 years and 7 years, with relapsing anti-NMDAR encephalitis; this included the frontal, parietal, temporal, and occipital cortex, and the thalamic nuclei. PET was reportedly more sensitive than MR imaging in these patients. However, a previous report of acute anti-NMDAR encephalitis contrasted this with signs of cortical hypermetabolism. An adolescent with anti-NMDAR encephalitis was found to have abnormal cerebral blood flow in on 99m Tc-labeled d,l -hexamethylpropylene amine oxime single-photon emission CT (HMPAO SPECT). Images showed multiple focal areas of radiotracer uptake in a corticobasal hyperperfusion pattern, which suggested the possibility of frontobasal circuitry involvement. Repeated brain 99m Tc-HMPAO SPECT showed almost complete normalization.
Electroencephalography
Electroencephalograms (EEGs) are consistently abnormal in most patients, and show nonspecific slowing and disorganization of background activity. These findings may be associated with electrographic seizures, which are more common in the initial disease stage. During the catatonic stage slow, continuous, delta, or theta, rhythmic activity predominates. The slow rhythmic activity has not been associated with movement abnormalities and has not responded to antiepileptic medication. VEEG monitoring may be beneficial in diagnosing seizures appropriately.
Cerebrospinal Fluid and Serum
NMDARs form part of the ligand-gated cation channels, which play an important role in synaptic transmission and plasticity. The NMDARs are heteromers of NR1 subunits that bind to glycine and NR2 (A, B, C, or D) subunits, which bind to glutamate. The presence of NMDAR antibodies was confirmed by serum or cerebrospinal fluid (CSF) reactivity with hippocampal rat brain, cell-surface labeling hippocampal neurons in culture, and reactive NR1- or NR2-transfected human embryonic kidney (HEK) cells. Definitive diagnosis of anti-NMDAR encephalitis is established by demonstrating antibodies to the NR1 subunit in patients’ serum or CSF. In a series of children with anti-NMDAR encephalitis, all patients had CSF or serum antibodies that reacted with extracellular epitopes of the NR1 subunit. Paired CSF and serum samples from 21 patients showed stronger antibody reactivity in CSF. Patients with a teratoma had higher antibody CSF titers than those without a teratoma. Seven patients treated with empiric plasma exchange or intravenous immunoglobulins had positive antibodies in CSF, but not in serum. Antibody titers may change over time. A patient who recovered showed a high serum antibody titer and absent or barely detectable CSF antibody titer at follow-up. Although viral encephalitis, stroke, epilepsy, and systemic lupus erythematosus have been associated with NMDAR antibodies, the NR1 receptor region appears to associate uniquely with anti-NMDAR encephalitis. This finding is consistent with an immune response, systemically triggered by a tumor or another unknown cause, which is reactivated and expanded in the central nervous system (CNS).
CSF white blood cells are often increased, in the range of fewer than 200/mm 3 , and pleocytosis was found in 87% of a series of children. Protein concentration is normal or mildly increased, and CSF-specific oligoclonal bands are positive in 60%. The immunoglobulin G (IgG) index and oligoclonal band testing are clinically useful, as they can be abnormal in the setting of normal CSF cell counts and protein concentrations.
Tumor Association
Approximately 80% of patients with anti-NMDAR encephalitis have so far been women. The detection of a tumor is dependent on age, gender, and ethnicity. Approximately 60% of patients have tumors proven to contain nervous tissue. In women, frequency of ovarian teratoma amounts to about 62%, whereas only 22% of men have associated tumors (testicular teratoma and small-cell lung cancer). In adolescent females younger than 18 and children younger than 14 years, tumors were diagnosed in 31% and 9%, respectively. The ovarian teratomas of 25 patients studied expressed NMDARs in each case. There is one case report of a child with anti-NMDAR encephalitis and neuroblastoma. Ovarian tumors were found in a few patients who had surgery with exploratory laparoscopies and blind oophorectomies, but in others no tumor was detected.
Pelvic or testicular ultrasonography is an appropriate initial screen for ovarian or testicular teratomas. MR imaging has been used as a more sensitive test for small ovarian tumors. Yearly screening for a tumor, particularly if the patient has a recurrence or continues to be symptomatic, is recommended.
Pathogenesis
The pathogenic role of antibodies in anti-NMDAR encephalitis can be established using the following in vivo and in vitro criteria: (1) if antigens are membrane proteins then antibodies should bind to extracellular antigenic epitopes in living cells and/or tissues; (2) the antibody’s ability to recognize an antigen should be assessed by expressing the antigen in heterologous cells, which are assayed by immunostaining or immunoprecipitation and Western blot; (3) antibodies cause structural or functional changes of the target antigen that can be established in vitro in dissociated neuron cultures and in vivo after antibody infusion, and antibody treatment may alter the target antigen leading to secondary change in cellular or synaptic function; (4) the clinical syndrome associated with the antibody should resemble the phenotype of the pharmacologic or genetic manipulation of the antigen; (5) passive transfer of the antibodies to animals should recreate the effects of the antibodies on the antigen as well as the clinical features of the disorder; and (6) cellular and synaptic changes, in addition to clinical signs and symptoms, should improve as the antibody titer is reduced.
The anti-NMDAR antibody fulfills these criteria. Antibodies of patients with anti-NMDAR encephalitis recognize the N-terminal extracellular domain of NR1 and bind, cap, cross-link, and internalize NMDARs. The presence of NMDAR antibodies was confirmed by serum or CSF reactivity with hippocampal tissue of rat brain, cell-surface labeled cultures of hippocampal neurons, and reactivity with NR1- or NR2-transfected HEK cells. This process caused a specific, titer-dependent, and reversible reduction of NMDAR surface and total cluster density, resulting in NMDAR-mediated synaptic dysfunction. Patients’ antibodies decreased NMDAR synaptic currents on whole-cell patch-clamp recordings of miniature excitatory postsynaptic currents in rat hippocampal neuron cultures, but did not change the localization or expression of synaptic proteins, other glutamate receptors, synapse number, dendritic spine complexity, or cell survival. The effects of NMDAR antibodies resemble those of the NMDAR antagonists phencyclidine and ketamine.
The various stages of the syndrome probably arise from an antibody-mediated progressive reduction of NMDAR clusters and function, followed by a restoration of receptor function during recovery. In vivo experiments have demonstrated that infusion of patients’ antibodies into rat hippocampus tissue substantially reduced NMDAR density, which was similar to the decrease of these receptors found in the hippocampi of autopsy specimens. The correlation between antibody titers and neurologic outcome and the reduction in number of postsynaptic NMDAR caused by the antibodies suggest that they play a role in the pathogenesis of the disease. Removing antibodies from the cultures increases the density of postsynaptic NMDAR and explains the potential reversibility of symptoms. Postmortem analysis of the hippocampi of 2 patients who died of this disorder showed a significant decrease in NMDAR density. The role of the immune response is further supported by the correlation between antibody titers and symptoms, and the frequent clinical response to immunotherapy. The reversibility of the disorder suggests immune-mediated neuronal dysfunction rather than irreversible degeneration.
Synaptic plasticity is thought to be an important mechanism for memory, learning, and cognition. These neurologic processes require glutaminergic synaptic localization and trafficking via NMDA and AMPA receptors. A suggested model of the disorder is that a decrease of NMDARs in inhibitory GABA neurons and glutamate synapses causes multiple effects, including disinhibition of excitatory pathways and increased extracellular glutamate. The clinical syndrome disinhibits the frontostriatal network, leading to symptoms of psychosis, catatonia, rigidity, dystonia, and mutism. The brainstem central-pattern generator is disinhibited, causing complex movement disorders, and the brainstem respiratory network is disinhibited, resulting in respiratory dysfunction.
The immunopathology, CSF pleocytosis, oligoclonal bands, and intrathecal synthesis of NMDAR antibodies suggests that the humoral immune also response plays an important role in the pathogenesis of this disorder. The tumor immunopathology suggests that the tumor may assist in triggering the anti-NMDAR immune response, likely contributing to the breaking of immune tolerance. However, this disorder can also present as a nonparaneoplastic, autoimmune syndrome. After initial systemic immune activation there is an expansion of the CNS immune response. Synaptic autoantibodies in the CNS may be the result of peripherally synthesized antibodies passively crossing a disrupted blood-brain barrier and intrathecal antibody synthesis by plasma cells.
The prodromal illness may cause transient disruption of the blood-brain barrier. After systemic immune activation by an NMDAR -expressing tumor or unknown factors, memory B cells are restimulated. These cells undergo antigen-driven affinity maturation, clonal expansion, and differentiation to become NMDAR antibody-secreting plasma cells. Cross-reactivity of antibodies against other antigens can occur if the epitopes are sufficiently similar. Symptoms of a viral prodrome occurred in 48% of children; however, a common virus was not found to suggest a postinfectious autoimmune process in patients without teratoma. A few patients had positive serology or direct demonstration of a systemic infection, preceding the symptoms of anti-NMDAR encephalitis. One out of 4 patients with neurologic complications, attributed to influenza H1N1, had NMDAR antibodies. This patient showed characteristic symptoms of anti-NMDAR encephalitis. A few patients with anti-NMDAR encephalitis had positive mycoplasma serology with negative CSF polymerase chain reaction. A child was reported with anti-NMDAR encephalitis after a booster vaccination against tetanus, diphtheria, pertussis, and poliomyelitis. These infections may act as an adjuvant, boosting the immune response. It is possible that a genetic predisposition in addition to a viral or neoplastic trigger may be necessary to initiate the disease. A 3-year-old child with anti-NMDAR encephalitis had a microdeletion of chromosome 6p that involved the human leukocyte antigen cluster, which suggested a predisposition to autoimmunity. A genetic predisposition to autoimmunity has been suggested in children, with antinuclear or thyroid peroxidase antibodies in addition to NMDAR antibodies.
Immunopathology
Postmortem examination of 2 adult patients who died of anti-NMDAR encephalitis showed microgliosis and IgG deposition in the hippocampus, forebrain, basal ganglia, and spinal cord. Anti-NMDAR antibodies were predominantly IgG1, and included IgG2 and IgG3 subtypes. No complement deposits were observed in the examined areas of the CNS. B cells (CD20) and plasma cells (CD79a) were predominant in the perivascular spaces. Cells expressing cytotoxic T-cell markers were rare or absent in the areas examined. In 14 patients with anti-NMDAR encephalitis, a brain biopsy was normal or nonspecific. Histology included perivascular lymphocytic cuffing with predominantly B cells or microglial activation, and sparse parenchymal T-cell infiltrates. Neuronophagic nodules were absent and viral assays were negative. The ovarian tumors of 2 patients who underwent postmortem examination and of 9 other patients with ovarian tumors who survived expressed neuronal antigens, and contained neurons that expressed NMDAR and reacted with patients’ antibodies. Nervous tissue was demonstrated by cell morphology and the presence of dendritic neuronal processes, using microtubule associated protein 2 (MAP-2) antibody. All tumors from patients with NMDAR antibodies had extensive T-lymphocyte infiltrates, macrophages or monocytes, B lymphocytes, and plasma cells.
Differential diagnosis
The differential diagnosis of anti-NMDAR encephalitis is broad, and typically includes subacute and chronic diffuse inflammatory disorders of the brain. When all the clinical features are present, the diagnosis can be entertained on a clinical basis, although confirmation by serum and CSF anti-NMDAR antibodies is essential. The differential diagnosis widens in the absence of all clinical features and the presence of atypical signs and symptoms. It is clear that with an expanding phenotype and accruing literature, certain specific features may become more helpful in the clinical suspicion of this condition.
Because some cases present with status epilepticus, acute infectious causes such as bacterial and viral infections of the brain need to be considered at presentation. Herpes simplex virus type 1 (HSV-1), human herpesvirus type 6, enterovirus, and mycoplasma are among the multitude of viruses that are possible. Subacute and chronic autoimmune-associated encephalitis, including pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections, Sydenham chorea, autoimmune synaptic receptor encephalitides including neuronal antibodies to GABA, AMPA, and LGI1 receptors, Hashimoto encephalopathy, Rasmussen encephalitis, and encephalitis lethargica, are some of the other conditions that may need consideration based on the clinical features. CNS vasculitis, either primary (primary CNS angiitis) or secondary (due to chronic infection, inflammatory processes), also must be considered in the differential diagnosis at presentation. A presentation similar to anti-NMDAR encephalitis has been reported in seronegative neuromyelitis optica. There are additional poorly understood conditions such as acute encephalitis with refractory repetitive partial seizures (AERRPS) and febrile infection–related epilepsy syndrome (FIRES) that may have overlapping clinical features. Primary psychiatric disorders need consideration if the clinical features are solely of psychiatric nature.
Finally, medication overuse and abuse are rare considerations such that ketamine, phencyclidine, and neuroleptics causing neuroleptic malignant syndrome might have to be considered in individual cases ( Table 1 ).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


