CHAPTER 10
Aortic Stenosis and Pulmonary Stenosis
Aortic Stenosis
Definition
Congenital aortic stenosis may be caused by a variety of lesions that obstruct outflow from the left ventricle. Aortic stenosis occurs in approximately 3% to 6% of neonates with congenital heart disease.1,2 In utero, aortic stenosis may be an isolated lesion; however, associated cardiac malformations are seen in approximately 30% of cases.2–7 These malformations most commonly include hypoplastic left heart syndrome, coarctation of the aorta, endocardial fibroelastosis, ventricular septal defects, pulmonary stenosis, and mitral stenosis.3–5,8,9 Congenital aortic stenosis can be classified into three types, depending on the site of obstruction: valvular, subvalvular, or supravalvular stenosis.
Types
Valvular stenosis is the most common type of aortic stenosis, occurring in 0.25 of 1000 live births and in 60% to 75% of patients with aortic stenosis.10–13 It results from an abnormal formation of the valve cusps. In pediatric patients with valvular stenosis, 10% to 15% have symptoms in infancy. These patients usually have a more severe form of stenosis than do those in whom symptoms develop later in childhood.3,14,15 Neonates with valvular aortic stenosis frequently have severe mitral stenosis, endocardial fibroelastosis, hypoplastic left heart syndrome, and coarctation of the aorta. Atrial and ventricular septal defects are also seen.16,17 A fetus with valvular aortic stenosis has an increased risk for becoming hydropic in utero.18,19 Congenital valvular stenosis is three to four times more common in males than in females.20
Subvalvular stenosis is considered the second most common form of aortic stenosis, occurring in 8% to 30% of patients with congenital left outflow track obstructions.21,22 As with valvular stenosis, males are affected two to three times as often as females.23 Unlike valvular stenosis, severe subvalvular stenosis resulting from a fixed lesion (i.e., a membrane or fibromuscular tunnel) is rarely seen in newborns or infants.22,24,25 It usually occurs later in childhood as a result of associated congenital heart defects. However, the dynamic form of this lesion has been reported in utero.26 Dynamic forms include the inherited disorders of asymmetric septal hypertrophy (ASH), idiopathic hypertrophic subaortic stenosis, and hypertrophic obstructive cardiomyopathy.27
Infants of diabetic mothers in whom the diabetes is not well controlled are often found to have a transient form of dynamic obstruction of the left ventricular outflow tract.28 Associated cardiac lesions are common in subvalvular stenosis, occurring in 50% to 65% of patients.23 They most commonly include ventricular septal defect, interruption of the aortic arch, and coarctation.29,30 Progressive subvalvular aortic stenosis can also lead to damage of the aortic valve, resulting in aortic regurgitation.31
It is also common to detect aortic stenosis caused by an obstructive cardiomyopathy in the recipient twin of a pregnancy affected with twin-twin transfusion syndrome (TTTS). In TTTS, the recipient twin’s heart is likely to become hypertrophic as a result of increased preload. As the septum and heart walls hypertrophy, they obstruct the left ventricular outflow tract, resulting in a stenosis.32 Pulmonary stenosis usually develops in these fetuses before the onset of aortic stenosis.
Supravalvular stenosis is the least common of the three types. It may be an isolated anomaly but is often associated with peripheral pulmonary stenosis.33 Supravalvular stenosis is described as a narrowing of the ascending aorta that may be localized or diffuse, originating at the superior margin of the sinuses of Valsalva just above the level of the coronary arteries.1,34
Three forms of supravalvular stenosis have been described. Fifty percent to 75% of patients have an “hourglass” deformity, with a constricting ridge at the superior margin of the sinuses of Valsalva caused by extreme thickening and disorganization of the aortic media.33,35
Approximately 25% of patients have a more diffuse type of narrowing extending along the ascending aorta.36
A discrete membrane above the valve has also been reported.35 In 1961, Williams et al37 described the association of supravalvular aortic stenosis, mental retardation, and elfin facies. This constellation of findings now bears the name “Williams syndrome,” which is a microdeletion of 7q11.23 involving the elastin gene.33,38 A total of 28% to 50% of patients with supravalvular aortic stenosis have Williams syndrome.39 The finding of supravalvular aortic stenosis along with peripheral pulmonary artery stenosis is nearly exclusive to patients with Williams syndrome.38 Neonates with Williams syndrome also have hypercalcemia, narrowing of peripheral systemic and pulmonary arteries, strabismus, auditory hyperacusis, gastrointestinal problems, and urinary tract abnormalities.1,33,38,40
Severe or critical aortic stenosis is associated with high mortality and morbidity rates. A critical stenosis is characterized by decreased left ventricular function and ductal dependence.41 Without intervention, critical aortic stenosis is likely to progress to hypoplastic left heart syndrome.42,43
Embryology
The etiology of congenital aortic stenosis is unknown but is thought to be multifactorial. De la Cruz et al44 and Grant45 have suggested that aortic valve stenosis may be the result of overgrowth of endocardial cushion tissue. Mechanical interference with blood flow, hypoxia, and pharmacological agents such as thalidomide have all been reported to produce experimental aortic stenosis or a hypertrophic left heart, or both.46–48 These factors presumably cause cellular death or abnormal differentiation of cells, thus interfering with normal blood flow through the developing heart. This reduction of flow results in underdevelopment of cardiac structures. Some forms of aortic stenosis are known to have an autosomal dominant transmission with variable expression, whereas other forms are thought to be spontaneous mutations.1
Occurrence Rate
Aortic stenosis has been reported to have a frequency of approximately 5% in live births.2,49 The suggested risk of occurrence for a fetus with an affected father is approximately 3%. If the mother is affected, the risk of occurrence in the fetus rises to 13% to 18%. The occurrence risk if a sibling is affected is approximately 2%; it is 6% if two siblings are affected.50
Sonographic Criteria
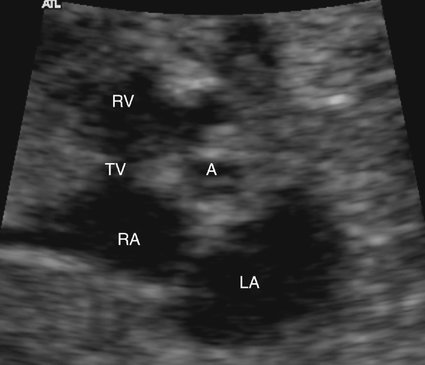
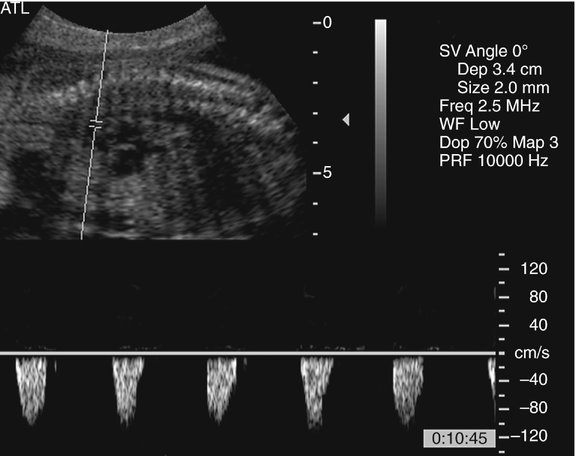
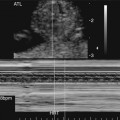
The prenatal diagnosis of aortic stenosis may be difficult, depending on the type of obstruction involved.50–52 In valvular stenosis, a structural abnormality of the aortic valves cusps is present. The aortic valve may appear thickened, immobile, fused or dysplastic (Fig. 10–1). The valve may also be bicuspid or unicuspid or in rare cases have more than three leaflets (Fig. 10–2). These findings can be extremely subtle in a fetus. Poststenotic dilation of the aorta may provide a clue to proximal stenosis. Pulsed and color Doppler imaging are essential in diagnosing stenosis. Flow distal to the aortic valve is increased and turbulent (Fig. 10–3); however, in some cases of critical aortic stenosis, no flow may be appreciated. Severe stenosis may lead to myocardial dysfunction and ventricular enlargement.34,53 In the setting of severe early-onset aortic stenosis, the decrease in blood flow is likely to result in a hypoplastic left ventricle.42,43
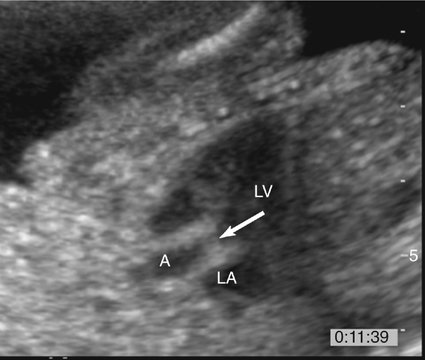
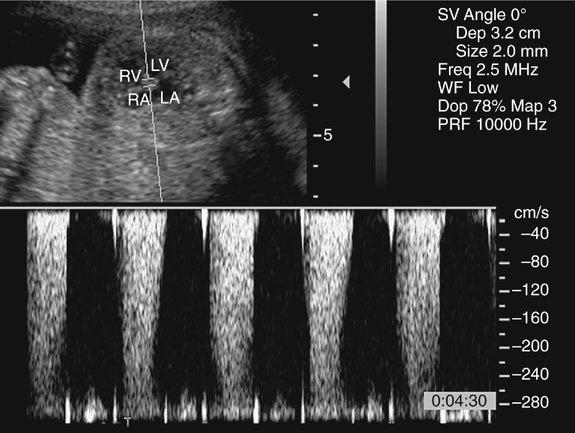
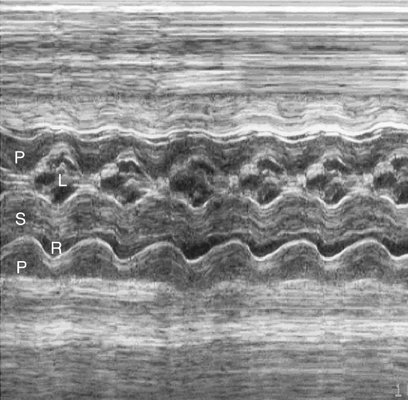
Endocardial fibroelastosis resulting from aortic valvular stenosis has been demonstrated prenatally.54 Sonographically, endocardial fibroelastosis appears as a hyperechoic rim around the affected chamber (Fig. 10–4). It is thought to result from blood stagnating within a chamber because of an outflow obstruction, causing fibrin deposits within the heart wall. Endocardial fibroelastosis has the potential to restrict chamber filling, hence impeding chamber growth.55 Retrograde flow in the aortic arch (Fig. 10–5) and left-to-right flow across the foremen ovale are also seen with critical aortic stenosis.42,43
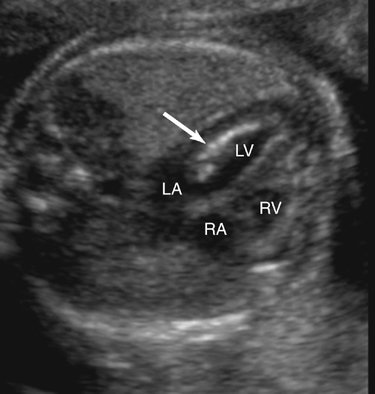
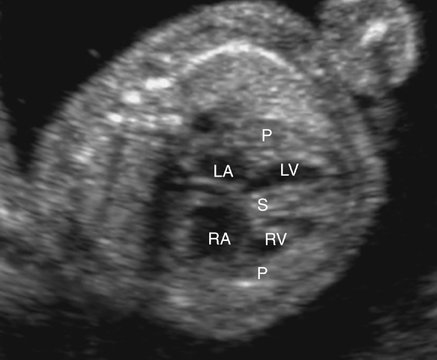
In the subvalvular form of stenosis, ASH or concentric hypertrophy of the left ventricle may be seen.56 The ventricular heart wall and the interventricular septum should be measured and compared with gestational age–related nomograms. In the case of ASH, which is often seen in fetuses of diabetic mothers, only the septum will be hypertrophied. With concentric hypertrophy, the interventricular septum and posterior heart walls will both be thickened, possibly reducing chamber size (Figs. 10–6 and 10–7). When ASH or concentric hypertrophy are identified in any fetal heart, serial fetal echocardiograms should be performed to detect the onset of valvular stenosis.
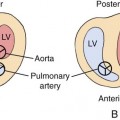
In all forms of aortic stenosis, the mitral, tricuspid, and pulmonic valves should also be investigated with pulsed or color Doppler imaging for evidence of stenosis or insufficiency. The aortic arch should be evaluated thoroughly because interruption of the aortic arch or coarctation is frequently associated with aortic stenosis. Although difficult in the fetus because of resolution limitations, an attempt should be made to image the aortic valve leaflets in a short-axis view to evaluate for bicuspid or unicuspid valve (see Fig. 10–2). A unicuspid unicommissural aortic valve is commonly seen in infants with aortic stenosis.57 Congestive heart failure may occur in utero; therefore signs of hydrops fetalis, such as pericardial or pleural effusions or ascites, should be investigated.
Treatment
Neonates with critical aortic stenosis display signs of circulatory collapse, cyanosis, and congestive heart failure. Without surgical intervention, the symptomatic neonate with severe aortic stenosis will not survive. Critically ill neonates require urgent intervention. At birth, prostaglandin E1 is usually given to maintain ductal patency, thereby relieving pulmonary hypertension and maintaining systemic perfusion.20,58 Surgical intervention for valvular aortic stenosis usually consists of balloon valvuloplasty, transventricular valvotomy, or open repair.2,29 Patch aortoplasty is used to treat supravalvar aortic stenosis, whereas fibrous membrane or muscular resections are performed with subvalvular stenosis. Typically, these types of surgeries are palliative. The majority of children with valvular stenosis will need valve replacements.2 Critical aortic stenosis requires eventual aortic valve or even aortic root replacement in many, if not all, cases.59,60
In cases of critical aortic stenosis, in utero balloon valvoplasty continues to be refined. Under ultrasound guidance, a cannula and needle are passed through the maternal abdomen, uterus, and fetal chest wall into the left ventricle. A guidewire and coronary balloon 10% are placed at the level of the aortic annulus.61 A balloon is then inflated within the aortic ring. Tierney et al42 reported 42 patients who underwent this procedure. Of the 30 cases evaluated (12 patients were lost to follow-up), aortic valvuloplasty was a technical success in 26 patients. Success was determined by a decrease in mitral regurgitation, bidirectional flow across the foramen ovale, and the return to antegrade flow in the aortic arch. Postnatally, eight achieved biventricular circulation and 18 had a functional single ventricle palliation. Significant aortic stenosis did not reoccur within the 26 successful procedures. Long-term outcomes of this relatively new procedure have not been established.
Prognosis
The prognosis in the various forms of congenital aortic stenosis varies with the severity of the obstruction and the associated abnormalities. Isolated or mild stenosis has a good prognosis. A mortality rate of 11% in the first 5 years of life has been reported. Surgical mortality rates for aortic valvotomy range from 9% to 67%.2 Rates can be higher depending on the severity of the stenosis, associated abnormalities. and the type of intervention necessary.14,29,61 With critical aortic stenosis, aortic valvuloplasty success rates are improving. However, the majority of fetuses will develop hypoplastic left heart syndrome in utero and require a Norwood procedure or heart transplant. In multiple studies, only a handful are born with a functioning biventricular heart. However, fetal hydrops was not identified in any of the live-born fetuses.42,43,53,61



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


