Abstract
Atelosteogenesis (AO) types I, II, and II are a group of usually lethal short-limb skeletal dysplasias. They are characterized by an abnormal, characteristic faces and hypoplastic or very shortened bones. All three forms have abnormal facies and short limbs, but associated findings, such as synpolydactyly, omphaloceles, and frontal encephaloceles, are seen in AO type I, multiple joint dislocations and abnormal hands in AO III, and increased nuchal thickness and hitchhiker thumbs in AOII help distinguish them from each other. AOI and AOIII are dominantly inherited; AOII is a recessive disorder. Neonates with all forms of AO frequently succumb usually in infancy resultant from respiratory compromise, although some individuals with AO III survive.
Keywords
atelosteogenesis, skeletal disorders
Introduction
Atelosteogenesis (AO) refers to a group of lethal short-limb skeletal dysplasias characterized by an abnormal, characteristic facies and hypoplastic or dysplastic tubular bones. Specific radiographic abnormalities, distinct histopathology, and differing inheritance patterns distinguish the three well-recognized types of AO. All three forms have abnormal facies and short limbs, but associated findings help distinguish them from each other; for example, synpolydactyly, omphalocele, and frontal encephalocele are seen in AO I, increased nuchal thickness and hitchhiker thumbs in AO II, and multiple joint dislocations and abnormal hands in AO III.
AO is caused by defects in genes involved in skeletal development. AO I and AO III result from heterozygosity for mutations in the gene coding for filamin B (FLNB). AO II, which is recessively inherited, is caused by mutations in the diastrophic dysplasia sulfate transporter ( DTDST, also referred to as SLC26A2 ) gene. Neonates with all forms of AO frequently succumb, usually in infancy, as a result of respiratory problems (pulmonary insufficiency, laryngeal hypoplasia, and recurrent infections), although some individuals with AO III survive into adulthood.
Disease
Definition
AO disorders are a group of chondrodysplasias with short limbs, unique forms of brachydactyly, and underdeveloped bones. The forms can also be distinguished based on cartilage growth plate histology. The diagnosis is suspected by skeletal findings, usually noted on routine prenatal ultrasound (US) and confirmed with molecular genetic testing, or postnatal radiology findings.
Prevalence and Epidemiology
AO I and III are autosomal dominant disorders, and germ line mosaicism has been reported. AO II is recessively inherited. Infants with AO I or AO II usually die in the immediate neonatal period from respiratory compromise. AO III is considered semilethal, but there have been long-term survivors.
Etiology and Pathophysiology
AO I and AO III result from mutations in the gene that encodes FLNB. Because these disorders are highly lethal, these mutations are sporadic in occurrence, although germ cell mosaicism has been seen. AO II is caused by either homozygosity or a compound heterozygosity for mutations in the diastrophic dysplasia sulfate transporter (DTDST, SLC26A2) .
Manifestations of Disease
Clinical Presentation
AO I/III disorders manifest with flattened facies, micrognathia, and shortening of all elements in the appendicular skeleton. Fetuses and neonates with AO I have a dysmorphic facies (hypertelorism, depressed nasal bridge, cleft palate, micrognathia), shortened trunk, rhizomelia, mesomelia, and brachydactyly with a distinct paddle-shape. Dislocations are common, particularly of the hips, and equinovarus is quite common. Neonatal death is due to respiratory insufficiency, usually from pulmonary hypoplasia and laryngeal hypoplasia. There is a spectrum of variability between AO I and AO III, and AO III is distinguished by better-developed long bones.
Fetuses and neonates with AO II have relative macrocephaly, midface hypoplasia, micrognathia, cleft palate, small chest with a protuberant abdomen, equinovarus deformity, and hitchhiker thumbs and toes, with a large gap between toes 1 and 2. The limbs are severely hypoplastic or dysplastic, with poor mineralization of the spine. Neonatal death is typically due to pulmonary hypoplasia and tracheobronchomalacia.
Imaging Technique and Findings
Ultrasound.
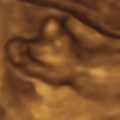
AO should be suspected in cases with very flattened facies with micrognathia and hypoplastic appendicular bones that are frequently absent or poorly formed, with hypoplastic distal ends, and polyhydramnios. Three-dimensional or four-dimensional US is useful in the evaluation of fetuses with skeletal dysplasias, especially for imaging the face and extremities ( Fig. 46.1 ).

AO I shows poor ossification of the spine, small chest, micromelia ( Figs. 46.2 and 46.3 ), abnormally shaped or bowing of the long bones, hypoplastic or dysplastic bones of the hands and feet (paddle shaped, Fig. 46.4 ), clubfeet, and severe facial dysmorphism (frontal bossing, micrognathia, and possibly cleft palate, Fig. 46.5 ). Distinctive features of AO II include hitchhiker thumbs and toes (see Fig. 46.1 ), equinovarus deformity of the feet with saddle gap deformity. The humeri and femora in AO II are severely slender at the distal ends. Phalanges are better ossified in AO II than in AO I and AO III.