- •
Size of the atrial septal defect
- •
Size of the ventricular septal defect (large = complete, small = transitional, none = incomplete)
- •
Valve attachments to the crest of the septum
- •
Presence or absence and degree of atrioventricular valve regurgitation
- •
Degree of balance or unbalance of the atrioventricular valve over the two ventricles
- •
Relative size of the ventricular cavities
- •
Presence or absence of left ventricular outflow tract obstruction (valve tissue obstructing or narrowed pathway due to small left ventricular outflow tract)
Anatomy and Anatomical Associations
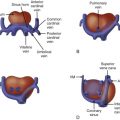
The term common atrioventricular canal defines a group of cardiac defects characterized by abnormal development of structures derived from the endocardial cushions during embryological development of the heart. Nomenclature varies depending on the school of thought and includes atrioventricular septal defect, endocardial cushion defect, and atrioventricularis communis; atrioventricular canal (AVC), our preference, is used for the purposes of this chapter. In AVC, there is absence of the atrioventricular (AV) portion of the septum as well as maldevelopment of the AV valves resulting in a common inlet to both the right and the left ventricles. The common AV valve is, by definition, abnormal and incompetence (regurgitation) frequently occurs. The defect in the AV septum can result in an exclusive atrial communication, a combined atrial septal defect (ASD) and ventricular septal defect (VSD), or an exclusive ventricular communication. The size and location of these defects dictates the clinical course. Fetal diagnosis of AVC is quite feasible and recognition in utero allows for prenatal counseling and possible identification of other noncardiac defects including chromosomal abnormalities (e.g., trisomy 21).
AVC is typically categorized based on level of septal shunting ( Figure 9-1 ). In all forms of the defect, the absent portion of the AV septum is the same; the septal communication is, therefore, determined by the position of the AV valve within the defect. The complete form of the defect is characterized by a large ASD and VSD with the common atrioventricular valve (CAVV) positioned between both defects. The atrial communication, known as an ostium primum defect, is located anteroinferior to the margin of the fossa ovalis and adjacent to the AV valves. Importantly, the septum primum portion of the atrial septum is intact. The VSD, known as a canal or inlet type defect, is situated in the posterior ventricular septum along the septal leaflet of the AV valve extending into the membranous septum. This location results in a “scooped out” appearance of the crest of the ventricular septum.

The CAVV anatomy includes a mural leaflet exclusively over the left ventricle and, typically, two leaflets exclusively over the right ventricle. The unique feature of AVC is that in all cases, two leaflets bridge the ventricular septum, one superiorly and one inferiorly. The superior bridging leaflet compromises the AV junction such that the aorta (wedged between the tricuspid and the mitral valve in the normal heart) sits more anterior than normal, resulting in the so-called gooseneck deformity. This abnormality results in foreshortening of the length of the left ventricle from the AV valve annulus to the apex and elongation of the length from the apex to the aortic valve annulus (these measures are equal in length in the normal heart). Patients with AVC are, therefore, at risk for the development of left ventricular outflow obstruction. The inferior leaflet always has chordal attachments to the ventricular septum. With regard to the superior bridging leaflet, these chordal attachments are more variable and are categorized by the Rastelli classification. In Rastelli type A, the superior bridging leaflet is divided at the level of the ventricular septum. Rastelli type A is associated with left ventricular outflow obstruction primarily because the attachments result in increased elongation of the left ventricular outflow tract. Rastelli B is quite rare; the superior leaflet is divided with attachments to a right ventricular papillary muscle that sits more superiorly on the ventricular septum. Type C is commonly associated with Down syndrome and is considered the most rudimentary of the classifications, with the superior leaflet being “free floating” with no attachments to the septum.
In addition to these classifications, the papillary muscle anatomy of the AV valve may be abnormal. They may be positioned closely or shifted counterclockwise in comparison with the normal heart. The presence of a solitary papillary muscle should be highlighted as a risk factor for repair and may reflect the potential for left ventricular inflow obstruction. In unusual cases of complete AVC, the CAVV is positioned superiorly attaching to the septum primum such that there is a large ventricular communication with no atrial level shunt.
The incomplete (also known as partial) form of AVC occurs when the chordae of the CAVV are densely adherent to the ventricular septum such that there is an ostium primum ASD but no VSD component ( Figure 9-2 ). Though there is a common, single AV valve annulus, there are two orifices. The superior and inferior bridging leaflets (as previously described) are attached to the ventricular septum by means of a connecting tongue of chordal tissue, which gives the valve the appearance of a “cleft.” Regurgitation through this cleft is quite common.

There is a transitional form of AVC as well, quite similar to the incomplete type. In transitional AVC, the chordal attachments are not quite as adherent to the ventricular septum resulting in a restrictive VSD or multiple small VSDs through the chordae.
The CAVV in AVC may be “balanced” over both ventricles or “unbalanced” such that the valve sits disproportionately over one ventricle more than the other. Typically, the contralateral ventricle is hypoplastic. Unbalance occurs in 10% of AVC cases. The more common right dominant type of unbalanced AVC is challenging to treat and often associated with other levels of left-sided obstruction such as subaortic stenosis and/or aortic arch obstruction. Right dominant unbalanced AVC can be quite marked, resulting in a variant of hypoplastic left heart syndrome. Left dominant type is less common and right ventricular outflow obstruction may occur.
Associated lesions seen with AVC include patent ductus arteriosus, ostium secundum ASD, coarctation of the aorta, and additional VSDs. Tetralogy of Fallot can also occur with AVC and is seen almost exclusively in patients with Down syndrome. The complete form of AVC with Rastelli C is most commonly associated with tetralogy of Fallot. AVC is often a component of heterotaxy syndrome and seen with other complex congenital heart lesions.
Frequency, Genetics, and Development
AVC occurs in approximately 34.8 per 100,000 live births but is more frequently present in fetal life. It is the ninth most common congenital heart lesion. Termination of pregnancy as well as fetal demise account for the higher prenatal incidence, although the actual number is not known.
There is a very strong association between AVC and Down syndrome. A child with Down syndrome is 1000 times more likely to have AVC than a child with normal chromosomes. The complete form of AVC is the most common seen in Down syndrome, although the other types can also be seen. In Down syndrome, other noncardiac anomalies can be seen including diffuse hypotonia, duodenal atresia, anal atresia, and Hirschsprung’s disease. After birth, many children with Down syndrome develop thyroid disease and/or a form of acute lymphocytic leukemia. Alzheimer’s disease can also occur in the fourth or fifth decade of life.
Recently, DSCAM (Down syndrome cell adhesion molecule) has been proposed as a possible candidate gene for the development of AVC. Familial clusters with different modes of inheritance have also been reported. AVC is also seen frequently in association with the heterotaxy syndrome, particularly the asplenia type.
Epithelial-to-mesenchymal transformation results in formation of the endocardial cushions (the primordia of the valves and septa). Mouse embryos that fail to develop these cushions experience demise late in gestation. The endocardial cushions are divided into superior, inferior, right, and left cushions. The AV canal septum and the AV valves are formed, at least in part, by the superior and inferior endocardial cushions. When the endocardial cushions fail to fuse, likely as a result of abnormal migration of mesenchymal cells, the canal portion of the atrial and ventricular septum do not develop and the tricuspid and mitral valves become a CAVV.
Fetal Physiology
The physiology of AVC has little impact on the fetal circulation unless there is significant CAVV regurgitation. The ostium primum ASD and the VSD are usually well tolerated in utero. Because the pulmonary vascular resistance is elevated in the fetus, there is little if any left-to-right shunting and, thus, no significant volume burden on the heart.
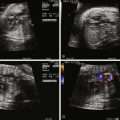
In contrast to many conotruncal abnormalities, the four-chamber view readily identifies the diagnosis; the AV communication can be seen with the single AV valve over the right and left ventricles. This is in contrast to the normal heart in which the triscupid valve annulus sits inferior to the mitral valve annulus. In balanced AVC, there is usually no discrepancy in size between the ventricles and ventricular shortening is typically normal.
In the short-axis view, the CAVV can often be seen en face with the superior and inferior bridging leaflets crossing into both ventricles. In the incomplete form, the ASD is sometimes difficult to see because a patent foramen ovale is also present. However, the short-axis view identifies the so-called cleft in the left component of the AV valve, which helps confirm the diagnosis. The distinction between incomplete and transitional AVC may be difficult in prenatal diagnosis because there may be minimal if any ventricular level shunting and the VSD may be too small to visualize by two-dimensional (2D) echocardiography.
Color Doppler imaging of balanced AVC typically demonstrates right-to-left shunting at the atrial level and bidirectional shunting at the ventricular level. Color Doppler imaging also readily identifies fetal AV valve regurgitation and its severity. The CAVV is, by definition, an abnormal valve. The mural leaflets may be hypoplastic and/or redundant; in the incomplete form, some element of regurgitation through the cleft is expected after birth. Because loading conditions are different in utero, AV valve regurgitation may not be present in the fetus. In fact, the presence of significant AV regurgitation before birth heralds a significantly abnormal valve, which may have a negative impact on outcome. If the AV valve regurgitation is severe, volume overload develops. Persistence of severe regurgitation often results in fetal compromise with heart failure and the development of hydrops fetalis.
Unbalanced AVC can also be identified prenatally. It should be suspected when AVC is diagnosed in association with ventricular size discrepancy. The severity of ventricular hypoplasia may progress during the pregnancy if there is only minimal inflow into one of the ventricles. When unbalanced AVC is recognized, distal obstruction from the hypoplastic ventricle should be suspected and searched for. In the right dominant type, the aorta is often significantly smaller (based on gestational age Z-scores) than the pulmonary artery. Aortic arch hypoplasia and/or coarctation of the aorta may be seen. Although a pressure gradient across the left ventricular outflow tract may not be observed because of the fetal physiology, direction of flow at the atrial septum can be helpful in determining the severity of disease. Left-to-right shunting at the atrial level signifies severe left-sided obstruction. After birth, these neonates generally require palliation with the Norwood procedure rather than a biventricular repair.
With left dominant type of unbalanced AVC, right ventricular outflow tract obstruction may be identified. In milder forms, the only evidence of pulmonary outflow obstruction may be that the pulmonary artery is smaller than the aorta (in the normal heart, it is larger). In more severe forms, there may be reversal of flow at the level of the ductus arteriosus with flow from the aorta to the pulmonary artery.
Complete heart block is sometimes seen in association with AVC in the fetus. It can occur with isolated AVC or in association with heterotaxy syndrome. The prognosis for this combination of defects with heart block is quite poor even with aggressive postnatal management.
Prenatal Management
Identification of AVC in utero may occur as early as the late part of the first trimester. Increased nuchal translucency, a screening tool for chromosomal and other anomalies of the fetus, can occur as early as 10 to 12 weeks’ gestation and can lead to early detection of congenital heart disease. If nuchal thickness is greater than the 95th percentile for fetal size, the risk of congenital heart disease increases from 8 to almost 50 per 1000 fetuses. Fetuses with Down syndrome will often have increased nuchal thickening as well. Early fetal echocardiography can be performed when there is suspicion based on nuchal translucency using transvaginal probes, particularly if the fetal position and/or acoustic window is inadequate for good visualization using the transabdominal approach.
The typical timing of prenatal diagnosis is at 18 to 20 weeks’ gestation. If the diagnosis of isolated AVC is made, amniocentesis should be strongly urged during counseling to rule out Down syndrome, because there is such a strong correlation. Families may consider termination of the pregnancy based on these findings. If the pregnancy is continued, monthly follow-up echocardiographic studies are suggested to follow progression of disease. In cases of balanced AVC with minimal or no AV valve regurgitation, the disease generally remains quiescent during gestation with no significant physiological impact on the fetus or the mother. For those with significant AV valve regurgitation, closer follow-up is warranted to assess for development of hydrops fetalis. When the AV valve regurgitation is severe, no good in utero treatment exists because this level of regurgitation generally signifies that the valve is structurally abnormal. Fetal demise from hydrops fetalis may occur and families should be counseled appropriately.
In unbalanced AVC, serial fetal echocardiography determines whether ventricular hypoplasia progresses. Direction of flow at the atrial septum and ductus arteriosus should be performed at each monthly assessment to help establish severity of disease. Serial studies allow for continued consultation with the family and discussion about changes that may have taken place.
The identification of an AVC on fetal imaging is an indication for a detailed obstetrical ultrasound evaluation as well as karyotype testing. From a cardiac standpoint, cesarean section is not necessary if a mother is carrying a fetus with AVC even if Down syndrome is present. Normal labor and delivery is well tolerated and is optimal for fetal transition. In general, fetuses with a diagnosis of AVC have a full-term gestation.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


