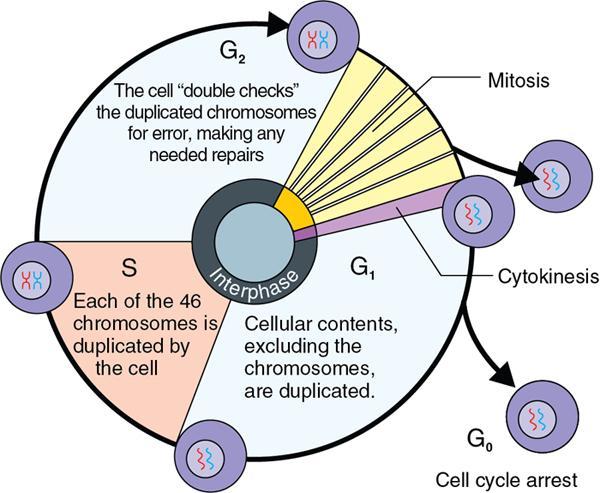
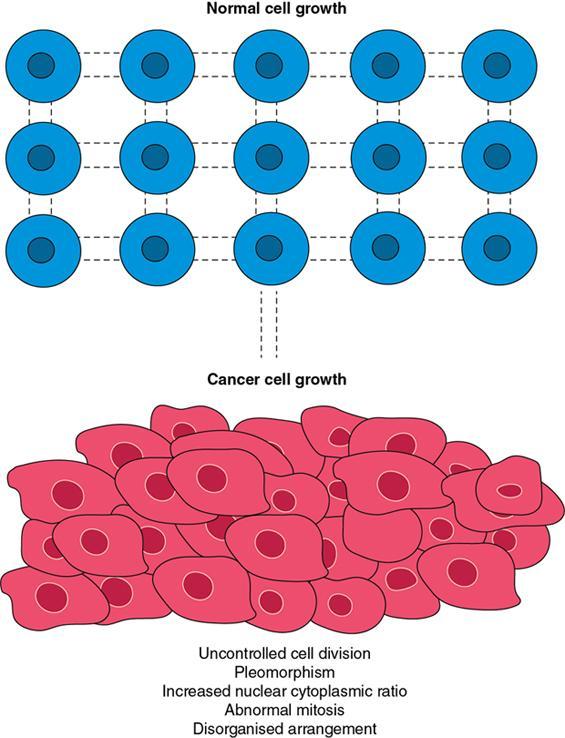
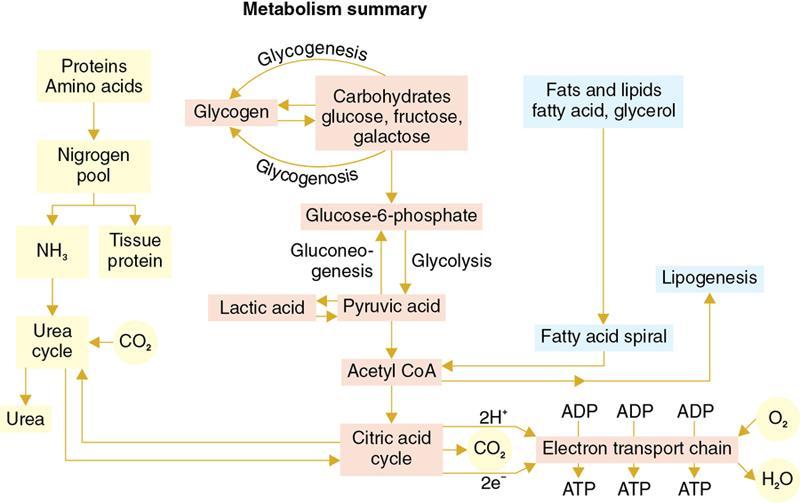
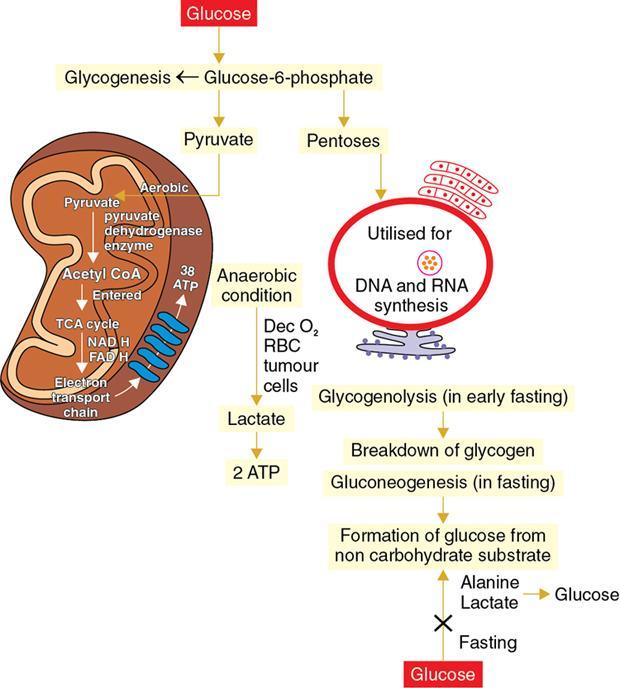
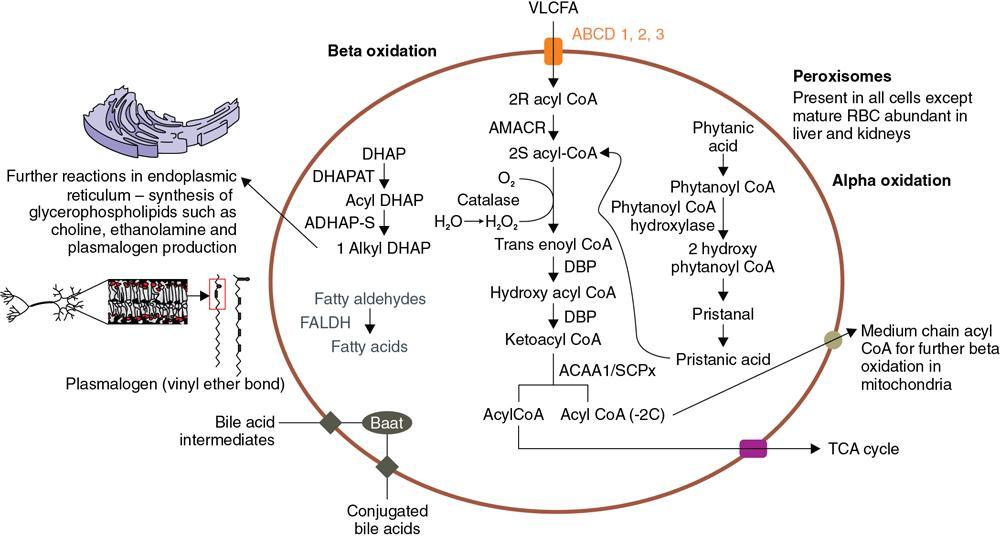
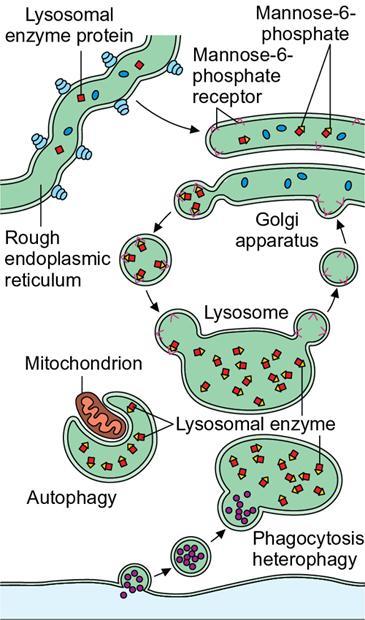
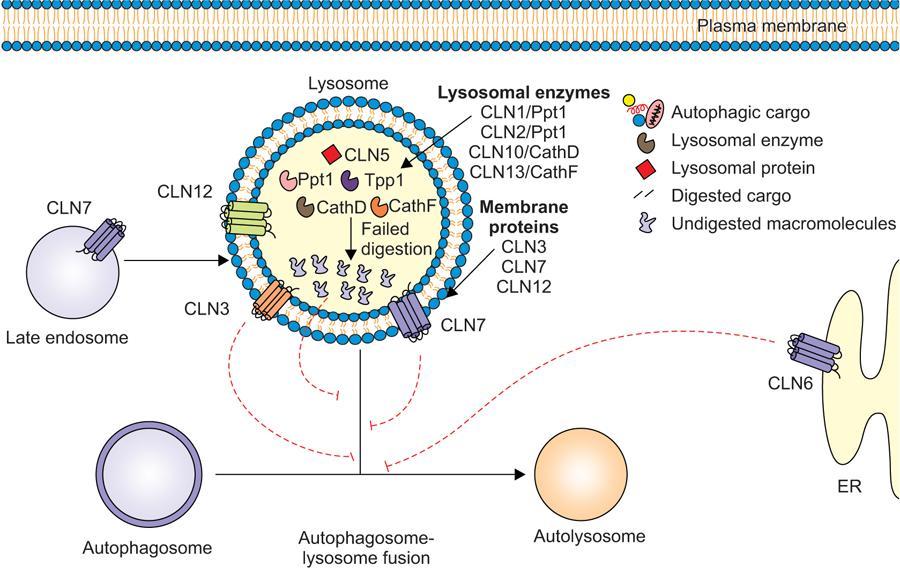
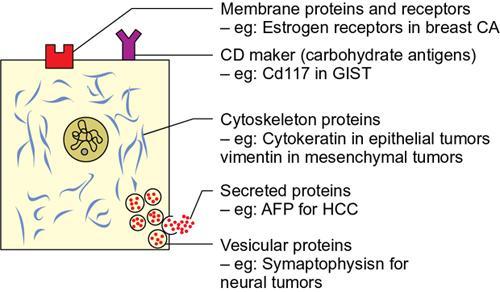
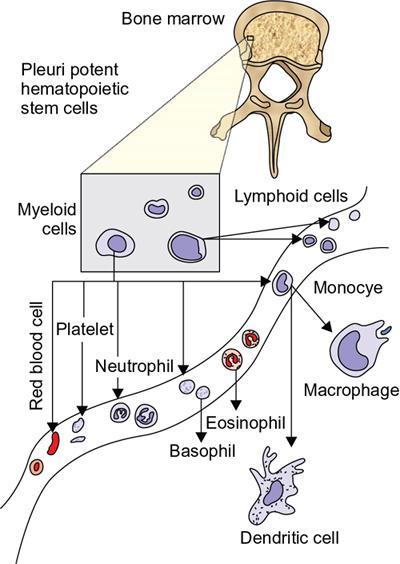
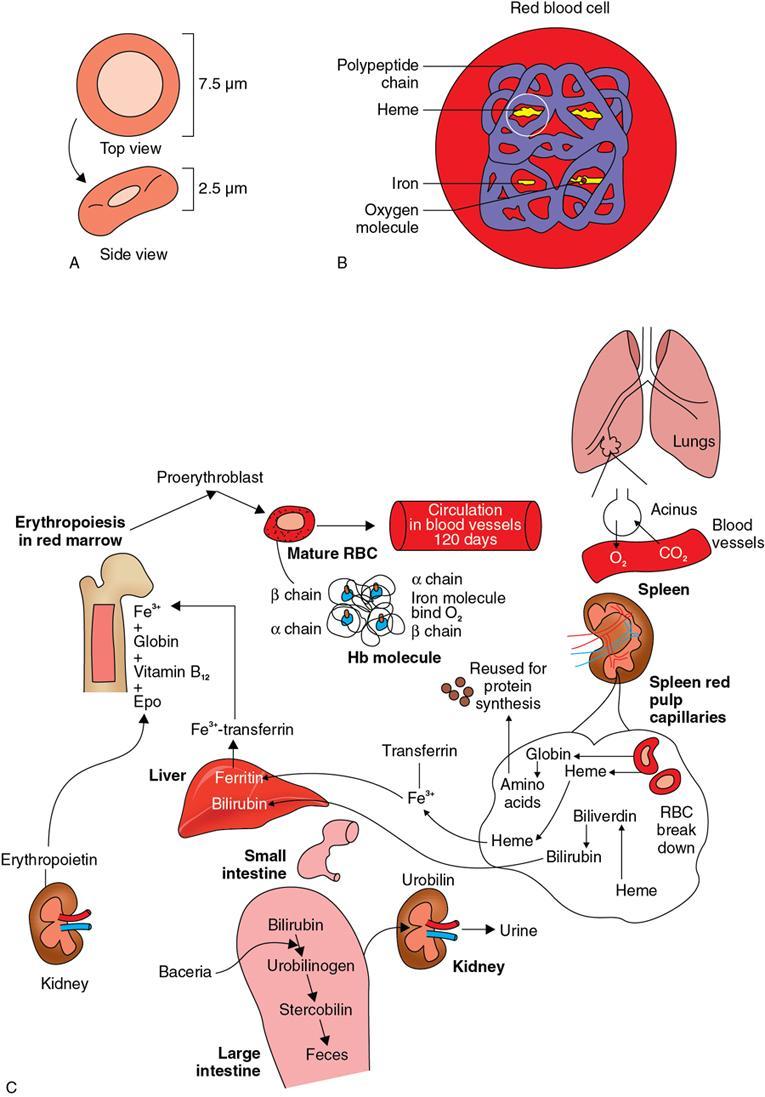
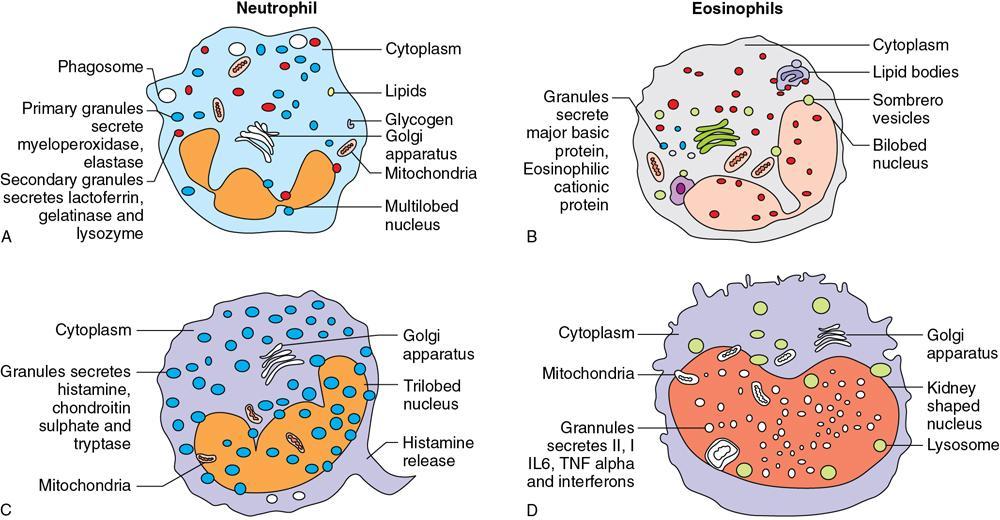
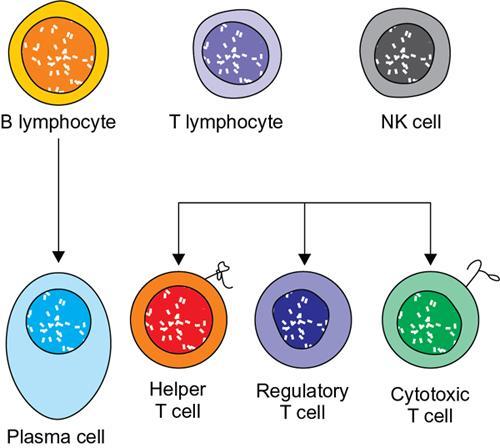
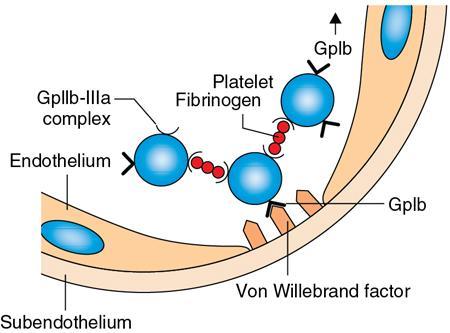
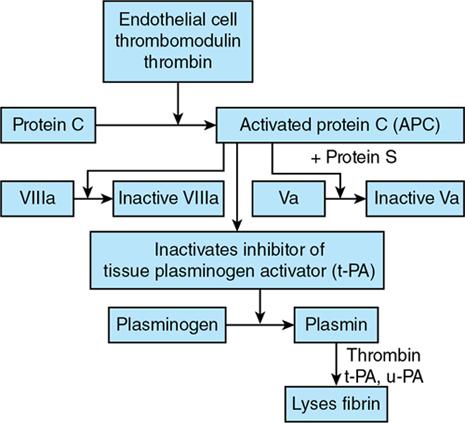
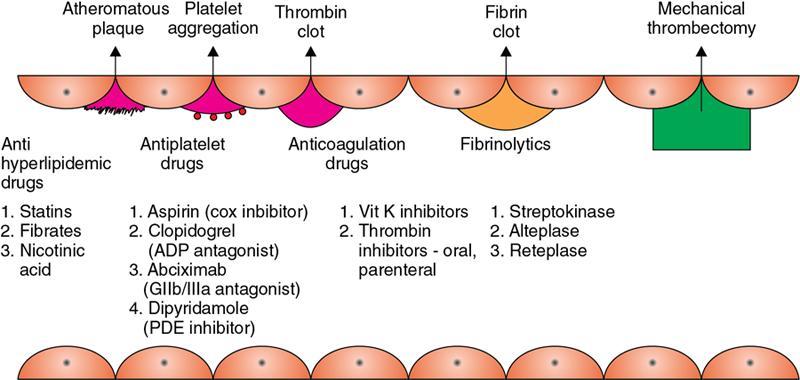
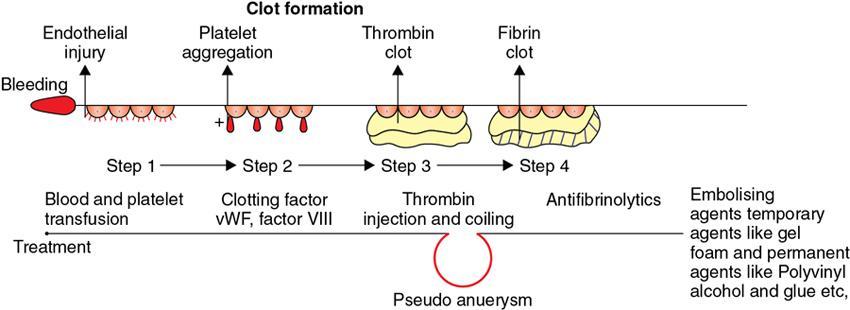
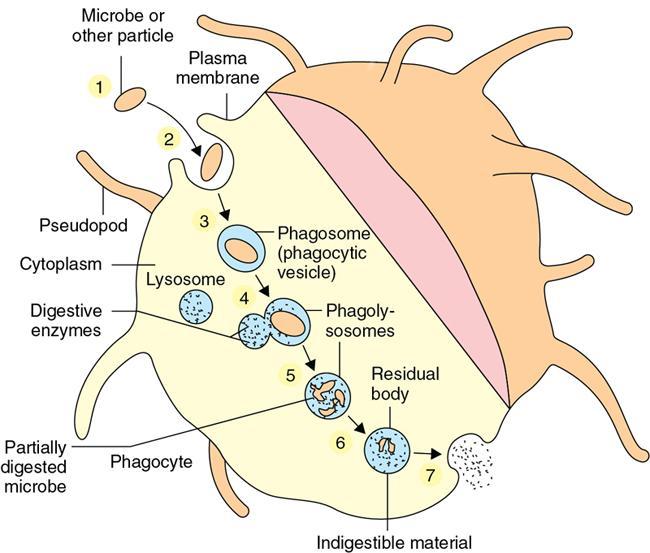
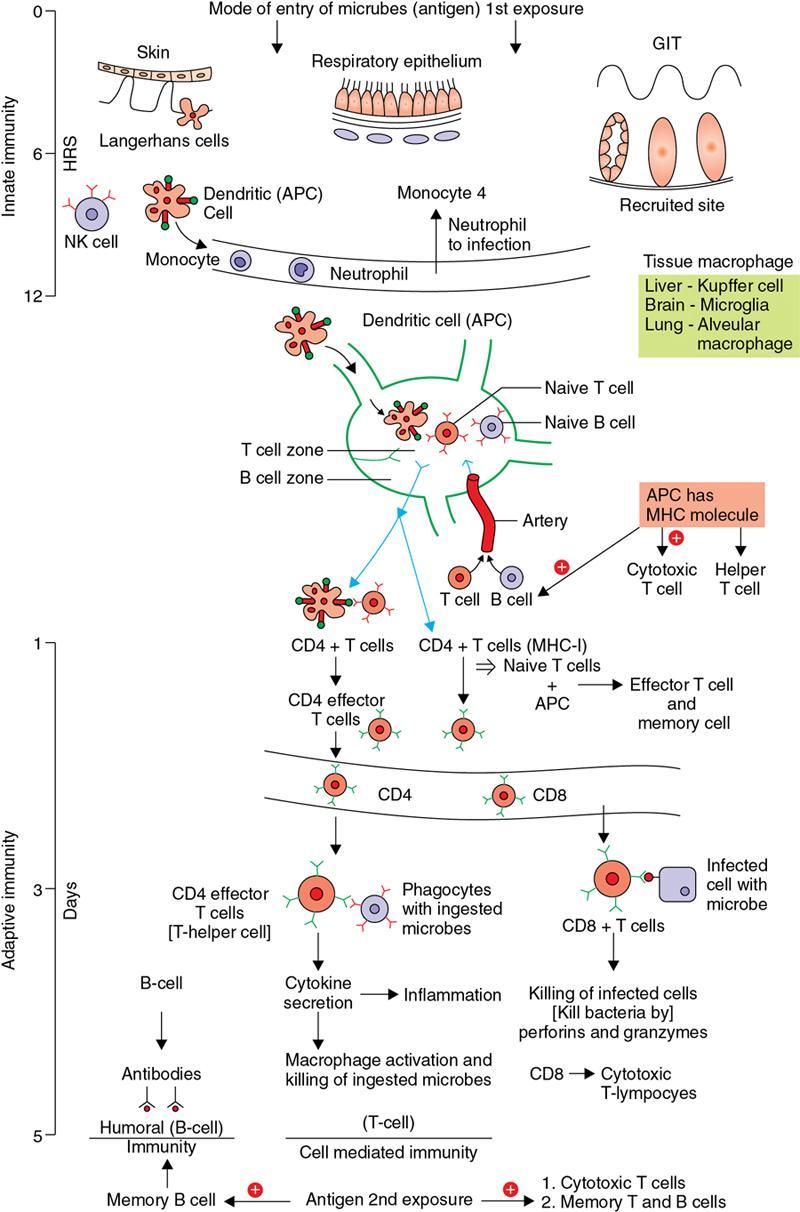
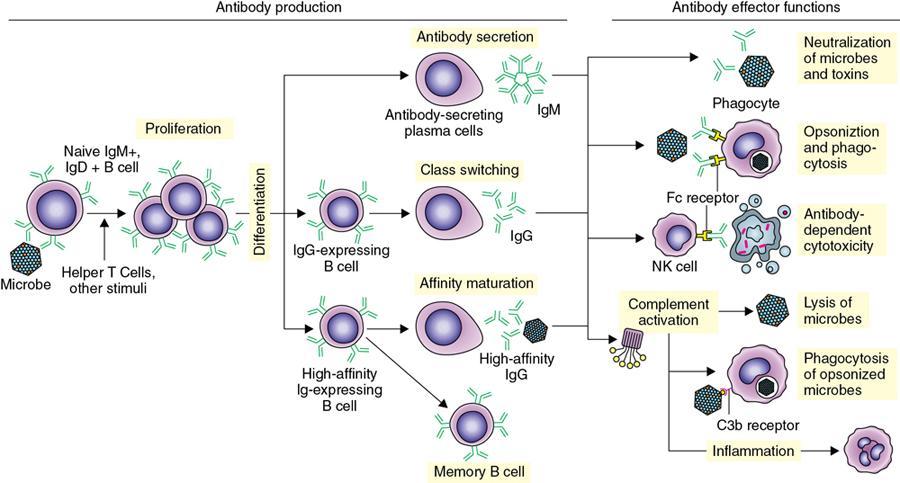
Amarnath C, Dhanalakshmi V., Gaurang Raval, Joel Kevin Raj Samuel, Menaka R., Sowmiya P. K. Radiology is one of the few broad specialities in the modern medical science which touches upon all other specialities in some way or other. To achieve the mastery of general knowledge of other specialities a radiologist must achieve mastery over basic medical sciences first. This would not only help to effectively communicate the observed abnormality but also help in guiding the further course of diagnosis or management. Why we must get back to our basic medical sciences in radiology: Radiology is the key diagnostic tool for many diseases and plays an important role in monitoring treatment and predicting outcome. Apart from macro-imaging, the use of imaging for ultrastructural diagnostics, nanotechnology, functional, quantitative diagnostics and molecular imaging has been steadily increasing. Radiologists are responsible for the implementation of these developments into the clinical setting and for ensuring the best use of these resources in patients care. For example, high resolution CT lung visualizes secondary pulmonary lobule and even alveoli – submillimetric micro-imaging of lung. Nowadays, the improved image precision and tissue differentiation has greatly increased the extent of diagnostic information and even demonstration of pathology is possible without the need of invasive tissue sampling. Formerly radiologists used to be trained only in the anatomy-pathology model and their correlation with images. The use of imaging for functional evaluation and cellular activity has created a new challenge where experience in physiology, cell function and other basic sciences are needed. The future of imaging will be functional and molecular imaging. Clinicians also require radiologists who understand the disease nature completely, keep updated with the most recent advances in disease process and have knowledge of relevant therapies. The advent of molecular imaging has in-turn changed a trend in medical imaging towards techniques aimed at tissue phenotype, its function, able to characterize cellular and molecular process, which will serve as a guide for new targeted therapies. For example, in cancer, the change in tissue metabolism and microenvironment and also the cellular expression of biomarkers have emerged as targets for imaging. Functional imaging with FDG PET can map tumour characteristics, such as hypoxia, vascularity, cellular proliferation that are known to impact the treatment outcome. In this chapter, we will look at the structure, function and organization of the cell, and the various metabolic processes which take place in it. Cells are the building blocks of the human body. There are approximately 100 trillion cells in our human body. The cell has nucleus and cytoplasm. The substances that make up the cell are collectively termed protoplasm which is mainly composed of five basic substances: water, electrolytes, proteins, lipids and carbohydrates. Cell Organelles: The cytoplasm is filled with both minute and large dispersed organelles and particles (Table 1.38.1, Fig. 1.38.1). The jelly-like fluid portion in which the organelles are dispersed is called cytosol. Cell cycle refers to the ordered sequence of events that take place in the cell in preparation for cell division. It consists of two gap phases (G1, G2), a synthetic phase (S) and a mitotic phase (Fig. 1.38.2). Rapidly dividing cells enter and leave the cell cycle frequently, whereas permanent cells are in a state of cell cycle arrest (Table 1.38.2). Fundamental physiologic changes that cancer cells show include the following: Self-sufficiency in growth signals: Tumours show the capacity to grow in the absence of external growth stimuli usually due to activating mutation of proto oncogenes. Insensitivity to growth inhibitory signals: Tumour cells do not respond to factors that inhibit proliferation of normal cells leading to uncontrolled growth. This is usually due to inactivation of tumour suppressor genes. Altered cellular metabolism: Tumour cells switch their metabolism to aerobic glycolysis to enable the synthesis of substrates and organelles needed for rapid cellular proliferation. This is known as the Warburg effect. Evasion of apoptosis: Tumour cells fail to undergo programmed cell death. Limitless replicative potential: Tumour cells like stem cells have no restriction for proliferation because they are resistant to undergo senescence and mitotic catastrophe. Sustained angiogenesis: Tumour cells induce angiogenesis themselves for their sustained growth. Ability to invade and metastasis: Tumour cells have the ability to invade locally and metastasis to distant sites. Ability to evade host immune responses: Tumour cells show number of alterations that help them to evade the host immune responses. The morphological changes in cancer cells include pleomorphism, large nucleus, increased mitoses, loss of polarity and anaplasia (Fig. 1.38.3). Pleomorphism is variation in size and shape of cells. Within the tumour cells are of various sizes and shapes. Large nucleus: Nucleus is large in size that nuclear–cytoplasmic ratio sometimes approaches 1:1 instead of normal 1:4 to 1:6. Increased mitoses: Number of mitoses is increased in cancer cells except in some well differentiated cancers. Loss of polarity: Orientation of cancer cells is disturbed in relation to the basement membrane. Anaplasia is lack of differentiation. The macromolecules important in cellular metabolism include carbohydrates, amino acids and lipids. Carbohydrates are energy substrate (fuel) of the body, whereas proteins are the building blocks of the body. Lipids are important for cell membrane and maintaining membrane fluidity. Even though these molecules perform various functions, all three can be useful for energy production (Fig. 1.38.4), carbohydrate in the well fed and fasting state, lipids and proteins can be useful as energy source in the fasting state. The final product in energy production is Adenosine Triphosphate (ATP). The total number of ATP produced in cellular respiration of one molecule of glucose is 30 ATP. ATP is the energy currency of the cell. It is needed for the maintenance of the normal function and activities of the cell. ATP plays an important role in the active transport of macromolecules across the cell membrane, ATP is useful in both intracellular and extracellular cell signalling. In CNS, adenosine modulates neural development, control of immune system and neuron signalling. ATP is also important for the muscle contraction; they bind myosin with actin. ATP is also needed for synthesis of RNA and DNA. Finally, ATP can be useful for structural maintenance of cell in the assembly of cytoskeleton structures. Glucose metabolism revolves around the breakdown of glucose into energy (Fig. 1.38.5). Glucose goes through various steps, initially being converted to pyruvate by glycolysis. Pyruvate can either be oxidized aerobically via the TCA cycle or be converted to lactate under anaerobic conditions. However, tumour cells anaerobically breakdown glucose to lactic acid even in the presence of oxygen. This phenomenon is called the Warburg Effect. Cancer cells have more demand of glucose, this is the basis for the use of FDG PET scan for diagnosis and staging of tumour. FDG uptake in tumours is proportional to the metabolic rate of viable tumour cells. Enzyme defects in glycolysis cause clinical disease (Table 1.38.3). Glycogen Storage Disorders (Glycogenosis and dextrinosis) are group of metabolic disorder caused by enzyme deficiencies affecting either glycogen synthesis, glycogen breakdown or glycolysis, typically in muscles and/or liver cells. Glycogen is a polysaccharide (meaning it is made up of multiple monosaccharide units of glucose) which is the primary storage form of carbohydrates in animal cells. Mucopolysaccharides, also known as glycosaminoglycans (GAGs), are polysaccharides consisting of repeating disaccharide units. Because GAGs are highly polar and attract water, they are used in the body as a lubricant or shock absorber. Its synthesis occurs in endoplasmic reticulum and Golgi apparatus. Degradation occurs in lysosome – Defect in degradative enzymes leads to storage disorder of mucopolysaccharides in lysosome, called mucopolysaccharidosis. Mitochondrial disorders are a group of disorders (Table 1.38.4) caused by defects in the mitochondrial DNA. Since all the mitochondria in a zygote are obtained from the ovum (as sperm does not contribute mitochondria), this group of disorders are maternally inherited. The lipids are a heterogeneous group of compounds, including fats, oils, steroids, waxes and related compounds. Lipids are classified as simple lipids (esters of fatty acids with alcohol, e.g. fat-oil, wax), complex lipids (phospholipids-glycerophospholipid, sphingophospholipid, glycolipid, etc.), derived lipids or precursor lipids (compounds derived from simple and complex lipids, they include fatty acids, glycerol, steroids, other alcohols, fatty aldehydes, ketone bodies, hydrocarbons, lipid-soluble vitamins and micronutrients, and hormones). Fatty acids (FA) occur in the body mainly as esters in natural fat and oil, free form can also occur in plasma in unesterified form as free fatty acids, a transport form in the plasma. Fatty acids can be classified depending on chain length as 1. Short chain fatty acid (C2–C6), 2. Medium chain fatty acid (C6–C14), 3. Long chain fatty acid (C15–C21), 4. Very long chain fatty acid (>C22). Depending on presence of double bond, FA can be classified as saturated fatty acid (no double bond) and unsaturated fatty acid (has double bond). Fatty acids are broken down into acetyl coA by oxidation which generates large amount of energy. Fatty acids are mainly used for energy in fasted states when glucose availability is less. But fatty acid oxidation is a main energy source for the heart, skeletal muscle and kidney even when glucose is ample. The various types of fatty acid oxidation are β oxidation, α oxidation, ω oxidation. Of these, the most common type of fatty acid oxidation is beta oxidation. Beta oxidation of long chain fatty acids, saturated fatty acid, unsaturated and odd chain fatty acid occurs in mitochondria. Beta oxidation of very long chain fatty acid occurs in peroxisomes and endoplasmic reticulum. Alpha oxidation occurs in peroxisomes and endoplasmic reticulum. ω oxidation occurs in microsomes (smooth endoplasmic reticulum). Peroxisomes contain multiple enzymes essential for normal growth, development and cellular metabolism. Peroxisomes are intracellular organelles that are involved in the oxidation (alpha and beta) of very long chain and monounsaturated fatty acids, bile acid synthesis, ether phospholipid biosynthesis and plasmalogen production, docahexanoic acid synthesis, glyoxylated detoxification (Fig. 1.38.6). Peroxisomes are present in all cells except mature RBC and are especially abundant in liver and kidneys. Peroxisomal enzymes are also involved in gluconeogenesis, lysine metabolism and glutaric acid metabolism. Alpha oxidation of fatty acids: Phytanic acid is the most common fatty acid to undergo alpha oxidation. It involves activation of phytanic acid, hydroxylation, removal of formylCoA, oxidation of pristanal, beta oxidation of pristanic acid. Beta oxidation occurs in peroxisomes and mitochondria. It differs in the enzyme involved and also FAs are completely degraded in mitochondria to CO2 and H2O, whereas peroxisomes can only chain-shorten fatty acids, which all need to be transferred further to mitochondria for complete oxidation. Inherited peroxisomal disorders can be classified in three groups: 1.due to deficiencies in peroxisomal formation, 2. disorders in which peroxisomes are formed but function improperly due to deficient function of single substrate transporter, 3. disorders in which peroxisomes are formed but function improperly due to deficient function of single enzyme. The abnormal fatty acids in peroxisomal disorders disturb the microenvironment of the cell causing dysfunction, atrophy and death of the cells. Impaired synthesis of plasmalogen also causes insufficiency and destruction of myelin. In Peroxisomal Biogenesis Disorders (PBD), peroxisomal organelles fail to form normally and are typically characterized by multiple enzyme deficiencies. Absent or reduced number of peroxisomes are present, with peroxisomal ghost (lack the normal components). PBD includes Zellweger spectrum disorder (includes Zellweger syndrome, x-linked adrenoleukodystrophy, infantile Refsum disease) and Rhizomelic chondrodysplasia punctata type 1 (RCDP) and other PEX7 conditions. PBD is characterized by abnormalities in neuronal migration, hypomyelination, germinal matrix injury and subependymal germinolytic cysts. Defective peroxisomes in oligodendrocytes also cause abnormal white matter formation and maintenance. Sjogren Larsson syndrome is rare autosomal recessive neurocutaneous disorder due to mutation in ALDH3A2 gene that causes deficiency of fatty acid aldehyde dehydrogenase (FALDH) enzyme. This enzyme is located in peroxisomes and microsomes and is responsible for oxidation of fatty aldehydes and alcohol into fatty acids. Therefore, fatty aldehydes, fatty alcohols and other related lipids get accumulated and disrupt the myelin. Triad of symptoms include congenital ichthyosis, spastic di-or quadriplegia, mental retardation. Neuroimaging reveals white matter disease and abnormal lipid peak on MRS. Lysosomes: Lysosomes are the key components for intracellular digestion (Fig. 1.38.7). They have hydrolytic enzymes called lysosomal enzymes or acid hydrolases, which are synthesized in endoplasmic reticulum and transported to the golgi apparatus. Within the golgi complex they undergo posttranslational modification including the attachment of mannose-6-phosphate which serves as an address label which are recognized by specific receptors. Thus, enzymes are targeted to lysosomes. The lysosomal enzymes catalyze the breakdown of variety of complex substances. Lysosomes are also involved in phagocytosis and in autophagy, i.e. turnover of intracellular organelles. Deficiency of lysosomal enzymes leads to accumulation of complex substances (inclusion bodies) which are normally degraded by lysosomes leading to lysosomal disorders. Defect in address label, i.e. mannose 6 phosphate, also leads to one form of lysosomal storage disorder (I cell disease). Defective autophagy also leads to secondary accumulation of autophagic substrates such as old, dysfunctional mitochondria and polyubiquitinated proteins. These accumulations trigger free radicals and causes cell damage and cell death. Neuronal ceroid lipofuscinosis is a neurodegenerative disease of childhood due to defective autophagy (Fig. 1.38.8). Ceroid lipofuscinosis neuronal proteins (CLN proteins) are located in the lysosomal matrix (CLN1, 2, 5, 10, 13) or at the lysosomal membrane (CLN3, 7). Disease-causing mutations in some of the CLN proteins inhibit autophagosome maturation (shown by dashed red lines) and autophagy. Mutated lysosomal hydrolases (CLN1, 2, 10, 13) are unable to degrade complex substrates which accumulate and impair lysosomal function. The lysosomal storage disorder includes glycogenosis, sphingolipidosis, sulfatidosis, mucopolysaccharidosis, mucolipidosis depending on the nature of accumulated metabolite. Of these, sphingolipidoses are a group of lysosomal storage disorder characterized by inherited deficiency of lysosomal hydrolase leading to intralysosomal accumulation of lipid substrates resulting from defective catabolism of the sphingolipids comprising cellular membranes. This group involves diseases such as Fabry disease, Farber disease, Gaucher disease and metachromatic leukodystrophy. Amino acids are building blocks of proteins. The chemical structure is composed of a carbon atom, an amino group, a carboxylic acid group and variable side chain termed as “R” which is common for all amino acids. Most amino acids are alpha amino acids. Non-alpha amino acids are beta alanine, beta amino isobutyrate, gamma amino isobutyrate. Depending on the dietary requirements, amino acids can be classified into essential and non-essential amino acids. Essential amino acids are those which cannot be synthesized by our body and should be provided through exogenous sources such as diet. The rest can be synthesized in the body through various pathways (Fig. 1.38.9, Table 1.38.5). Phenylketonuria, alkaptonuria, tyrosinemias, pheochromocytoma, albinism Defect in glycine cleavage system can cause hyperoxaluria, vitamin B6 deficiency, vitamin C toxicity, ethylene glycol poisoning Glycine shows an MR spectroscopy peak (at 3.55) in central neurocytoma Homocystinuria, vitamin B12/folate deficiency, vitamin B6 deficiency, oasthouse syndrome, cystathioninuria, cystinosis Hartnup’s disease, Drummond syndrome, carcinoid tumours Maple syrup urine disease The end product of the metabolism of the nitrogenous group of amino acids is ammonia. Ammonia is toxic to the cells and the brain in high concentrations. This ammonia is converted into urea in the liver by a process called the urea cycle for safe disposal by the kidneys (Fig. 1.38.10). Hepatic diseases or portosystemic anastomoses or collaterals can lead to hepatic encephalopathy. Defects of the enzymes of the urea cycle can lead to various disorders (Table 1.38.6). Common radiological manifestations include hyperintensities in basal ganglia and insular cortex in T1-weighted images. MR Spectroscopy – decreased NAA and myo-inositol peak, increased glutamate and glutamine. All living organisms have genetic information that is passed down from parents to offspring. In humans, this genetic information is located in the nucleus of every cell, in the form of a DNA molecule. This DNA molecule is organized into a structure called a chromosome. Each chromosome has a long (q) and a short (p) arm which are connected by a centromere. Humans have 23 pairs of chromosomes (i.e. 46 chromosomes) in somatic cells and 23 chromosomes in sperm and ova. The last pair of chromosomes are called sex chromosomes and consist of the XX chromosomes in females and the XY chromosome in males. Nucleotides are the repeating units which make up DNA. A gene is a segment of DNA which codes for a specific protein. There are around 25,000 genes in the human cell. DNA is organized into a compact form with the help of proteins called histones. This structure consisting of eight histones and two turns of DNA is called a nucleosome. Multiple nucleosomes form chromatid which is packaged to fit within the nucleus of the cell in a chromosome (Fig. 1.38.11). Nucleotides are macromolecules which consist of a nitrogenous base, a five-carbon sugar (ribose or deoxyribose) and a phosphate group. There are five nitrogenous bases seen in the human body – adenine, guanine (Purines) and cytosine, uracil, thiamine (Pyrimidines). In DNA, the adenine pairs to thiamine and guanine pairs with cytosine. These nitrogenous bases are metabolized to Xanthine and finally to uric acid. DNA is a large molecule which carries the genetic instruction in all living things in the form of genes which are organized into chromosomes. This instruction is used for protein synthesis. First, the information in a DNA is converted into mRNA by RNA polymerase (transcription). This mRNA is then converted into a protein by ribosomes (translation) (Fig. 1.38.12). During cell division, DNA also undergoes replication to pass on the genetic instruction to the daughter cells. RNA also consists of nucleotides, but it performs vastly different functions (Table 1.38.7). There are three different types of RNA (Fig. 1.38.13). There are multiple differences between DNA and RNA, the most important being that RNA uses Uracil instead of Thymine, and ribose instead of deoxyribose (Table 1.38.8). Defects in genes are a common cause for the pathogenesis of various diseases (Fig. 1.38.14). Many metabolic diseases are caused by defects in the genes coding for enzymes. Various other diseases (like sickle cell anaemia) are caused by defects in the genes coding for various proteins. Another important aspect is the role of various genes in carcinogenesis (Table 1.38.9) – various genes are mutated in cancer, including those involved in growth (oncogenes) and those involved in regulation (tumour suppressor genes). Tumour Tissue Markers: Immunohistochemistry is a technique used to investigate the tissue of origin of tumour cells. It is based on the principle that, different tissues express different proteins, based on their function. By staining tissues for these proteins, we can deduce the nature of the tissue under study (Fig. 1.38.15). Secreted (serum markers) proteins are covered later in this chapter. Blood circulates in the body and delivers essential substances like oxygen and nutrients to the body cells. It also transports metabolic waste products from different part of the body to the kidney. Two basic components of blood are fluid and cellular components. Fluid component is the plasma, cellular components are red blood cells, white blood cells and platelets. Haematopoiesis is the process of formation and maturation of blood cells from pluripotent haematopoietic stem cells in the bone marrow (Fig. 1.38.16). Haematopoiesis normally occurs in the marrow of long bones, the ribs and the vertebrae of the adult, while the principal sites of haematopoiesis in fetus are the yolk sac, spleen and liver. Red Blood Cells: RBCs are non-nucleated cells. They do not contain DNA, mitochondria, Golgi apparatus and insulin receptors. Pigment present in RBC is known as Haemoglobin. Energy is produced from anerobic glycolytic process. It is bi-concave disc shape, size is 7.2 micron in diameter. Erythropoiesis is a process of formation and maturation of red blood cells. Erythropoietin is a glycoprotein hormone that regulates erythropoiesis. Receptor involved in erythropoietin secretion is tyrosine kinase JAK/STAT, it inhibits apoptosis of RBCs. Kidney is the main source of erythropoietin. Life span of RBC is 120 days and determined by radio isotope method (iron, chromium). Fate of older cells – destruction occurs in capillaries of red pulp of spleen. From fragmented RBCs haemoglobin is released and phagocytosed by macrophage. Main functions of red blood cells are it carries oxygen from lungs to tissue, carries carbon dioxide from tissues to lungs, buffering action in blood, also helps in blood group determination by antigens (Fig. 1.38.17). When the primary sites of haematopoiesis in the adult fails, in conditions such as myelofibrosis, thalassemia and sickle cell the extramedullary sites are taken over for blood formation. Common sites of extramedullary haematopoiesis include liver, the spleen and the paraspinal regions. In addition to these common sites, the process can involve any organ. Identifying an extramedullary haematopoiesis is important, to exclude a neoplasm and alter the management and prognosis. Features of extramedullary haematopoiesis are splenomegaly, hepatomegaly, widening of medullary spaces and thinning of inner and outer table of calvarium, hair on end appearance of calvarium. In patients with various etiology of hemolytic anaemia are investigated by using Cr 51 (chromium 51) survival and sequestration technique. Cr 51 is used in assessing red cell survival, GI bleed and in cardiac ventriculography. Tc 99m RBC scan localizes the region of active blood loss of less than or equal to 0.1 mL/min at areas of radiolabelled material pooling. It is also used in detection of haemangioma. Heat-damaged RBCs labelled with Tc99m can be used to assess the splenic function in post-splenectomy, asplenia cases. Ultrasound beam from RBC gives rise to echo signals from blood for Doppler application. Increased prominence of internal echoes in lower shear rate venous blood flow compared with those of higher shear rate arterial blood flow is explained by red cell aggregation mechanism for blood flow echogenicity. In polycythemia false-positive hyperdense MCA sign is seen in CT brain. Back scattering of flow voids refers to a signal loss occurring with blood and other fluids like CSF, urine, moving at sufficient velocity relative to the MRI apparatus. The magnetic properties of Hb derivatives (oxy Hb, deoxy Hb, intracellular met Hb, extracellular metHb, haemosiderin) provide the dominant mechanism of MRI contrast in various patterns of haematomas like hyperacute, acute, early subacute, late subacute and chronic. Perfusion MRI detects different parameters, such as blood volume, cerebral blood, mean transit time and time to peak. Functional MRI based on blood oxygen level-dependent imaging (BOLD). Presence of deoxyhaemoglobin causes signal drop on T2-weighted image or T2 star weighted images because of deoxyhaemoglobin is paramagnetic while the oxyhaemoglobin is diamagnetic relative to surrounding tissue. Vascular endothelial growth factor (VEGF) is essential for tumour angiogenesis, this shows increased enhancement and leakiness in contrast studies. Sickle cell anaemia is caused by point mutation in beta globulin sixth codon replacement of glutamate residue with a valine residue. Sickle cell anaemia is diagnosed by sickling test by meta bisulphate, Hb electrophoresis, prenatal diagnosis by analysis of fetal DNA. MRI is used to monitor the effects of sickle cell disease such as musculoskeletal and neurological complication. Radio contrast agents are contraindicated in sickle cell disease because their high osmolarity may induce osmotic shrinkage of RBCs, impairs blood flow through the microcirculation, precipitate or exacerbate sickle cell crisis. Upconversion nano particles-coated RBCs are used for tumour imaging using MRI and PET. White Blood Cell: White blood cells (leucocytes) are Granulocytes, Monocytes and Lymphocytes. Granulocytes are classified into neutrophils, basophils and eosinophils. Monocytes are classified into macrophages and dendritic cells and lymphocytes are classified into B lymphocytes and T lymphocytes. Granulocytes are which have the ability to ingest viruses, bacteria and other parasites. They account for approximately 60% of WBCs. The sub-types of granulocytes are neutrophil, basophil and eosinophil. Neutrophils are a part of the innate immune system and an essential line of defence against bacteria. They are also known as “band neutrophils” (Fig. 1.38.18A). Basophils secrete histamine responsible for acute hypersensitivity reaction by causing vascular changes and increasing the capillary permeability. Basophils have IgE receptors, which also help them to produce hypersensivity responses (Fig. 1.38.18C). Mast cells is a large tissue cell resembling the basophil. It is present in bone marrow and around the cutaneous blood vessels but does not enter the circulation. It secretes heparin, histamine and serotonin. Eosinophils have the ability to release toxic product (major basic protein) from their granules for killing pathogens, such as parasites and worms. They are easily recognized in stained smears by their large granules (Fig. 1.38.18B). Monocytes stimulate osteoclasts cells, which have the ability to dissolve bone. They are the largest type of WBCs (Fig. 1.38.18D). Two types of cells developed from monocytes are macrophages and dendritic cells. Macrophages are phagocyte cells which eat any type of dead cell in the body. They are larger and live longer than neutrophils and have a large-size single nucleus that is often kidney-shaped. They are also able to act as antigen-presenting cells and tissue macrophages (e.g. histiocytes in connective tissues, Kupffer cells in liver, Langerhans cells in skin, osteoclast in bone, microglia in brain, alveolar macrophages in lungs). Dendritic cells are immature cells that aid the development of antigen immunity. The shape of the nucleus is small and round, which as the cell matures, turns into a large nucleus with an irregular star shape and cytoplasmic protrusions (dendrites). Dendritic cells are present in blood, bone marrow and also present as Langerhans cells in skin, interstitial cells in the lung and inter-digitating cells in thymic medulla. B lymphocytes (B-cells) mediate humoral immunity and can differentiate into a plasma cell. B cells mature in the bone marrow. T lymphocytes (T-cells) produce proteins called cytokines which help to direct the response of other cells (Fig. 1.38.19). Autologous white blood cells are radiolabelled ex-vivo using 99mTc-Hexamethyl propylene amine oxime (HMPAO) or Indium-111 oxime labelled. WBC scintigraphy allows additional information to be obtained regarding the degree of activity of disease and its extent in inflammatory bowel disease. WBC scan is used for evaluation of unknown site of infection in patients with high pre-test probability of infection, evaluation of extent of disease, soft-tissue infections and post op infections. Radiolabelled WBC scintigraphy accurately detects infective endocarditis and extra-cardiac septic embolisms. Radiolabelled WBC is not valid for the diagnosis of vertebral infections because of the frequent lack of uptake in the site of infection. FDG PET/CT in association with MRI provides the highest diagnostic accuracy. WBC scintigraphy combined with Tc 99-sulphur colloid used for prosthetic joint infection, whereas areas that concentrate leukocytes but not sulphur colloid indicate areas of infection. Platelets and Haemostasis: Platelets are disc shaped, absence nucleus with a diameter of 2.5 micron, with a life span of 7–10 days. They are involved in primary haemostasis. It constricts blood vessels and causes vascular spasm in broken blood vessels, form temporary platelet plug to stop bleeding, dissolve blood clots when they are no longer needed. During injury, endothelial damage occurs and transient vasoconstriction happens via endothelin and neural stimulation, vWF binds to exposed collagen. Platelets bind on von Willebrand factor (vWF) via Gp1b receptor at the site of injury – undergo conformational change and release adenosine diphosphate (ADP), calcium, thromboxane A2 (TXA2), ADP helps platelets bind to endothelium. ADP binds to P2Y12 receptor, induces Gp IIb/IIIa expression at platelet surface and platelet gets activated. Fibrinogen binds to Gp IIb/IIIa and links platelets finally form the primary haemostatic plug (Fig. 1.38.20). Activation of clotting factor and coagulation of blood is called secondary haemostatic plug. Blood coagulation occurs in three major stages. Stage 1: activation of Stuart–Prower factor by intrinsic and extrinsic pathway, stage 2: Formation of thrombin from prothrombin, stage 3: Formation of fibrin from fibrinogen. Regulators of coagulation: Thrombomodulin activates the protein C and protein S, also inactivates factor Va and V111a leads to inhibition of intrinsic mechanism of clotting. Plasmin inhibitors and plasminogen activator inhibitors are inhibitor of fibrinolysis. Plasminogen activator inhibitors inhibit tissue type (tPA) and urokinase type (uPA) plasminogen activator, alpha 2 macroglobulin and alpha 2 antiplasmin are inhibits plasmin. When mutation occurs in prothrombotic or antithrombotic agents in blood cause stroke in young, e.g. factor V mutation (Leiden mutation), protein C and S mutation (Fig. 1.38.21). Indium 111 oxime labelled platelets are used to assess platelet survival and localization of active thrombosis. Scintigraphy provides best results in case in which the platelets are already radiolabelled at the onset of thrombosis, and provide information on intracardiac thrombi. Disorders of Bleeding and Coagulation: It can be categorized as disorders of platelet, coagulation, due to excessive fibrinolysis, vascular defects (Table 1.38.10). Appropriate precautions are to be taken before performing diagnostic/therapeutic intervention procedures in patients with platelet function disorders. Thrombotic thrombocytopenic purpura is a congenital or acquired deficiency of ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) activity which produces thrombocytopenia by disorganized platelet clumping and aggregation. Heparin-induced thrombocytopenia is an immune-mediated reaction to heparin with decreased platelet count of less than 100,000 or >50% from baseline. Clinical suspicion should lead to discontinuation of heparin and change to direct thrombin inhibitor. Imaging findings include end organ thrombosis, venous and arterial thromboembolism, cerebral stroke, myocardial infarction, pulmonary embolism, aortic and mesenteric thrombosis, solid organ infarcts and DVT. The importance of identifying accessory splenic tissue is of utmost importance for satisfactory post-splenectomy results of idiopathic thrombocytopenic purpura (ITP). The recurrence of thrombocytopenia post-splenectomy is an indication for scintigraphy TC 99m nanocolloid/sulphur colloid/heat denatured RBCs (better detection rate as compared to CT). Stages of thrombus formation and their treatment options (Fig. 1.38.22): Mechanism of clot formation and therapeutic options (Fig. 1.38.23): The immune system is a complex network of cells and proteins that defends the body against infection. The organs of immune system are classified as primary lymphoid organs which include bone marrow-dependent B-cells, thymus-dependent T-cells. Secondary lymphoid organs include lymph nodes, spleen, MALT (Mucosa-Associated Lymphoid Tissue located in the respiratory tract and GIT), tonsils and Peyer patches. There are two types of immunity. They are innate immunity and acquired immunity. Innate immunity is non-specific and present since birth. Adaptive immunity is acquired after antigen exposure and is more specific. Antigen-presenting cells (APCs) are types of cells, which are present in epithelia and most of the tissues, capture microbes and other antigens, process and present them to T cells. Professional APC – dendritic cells, macrophage and B cells. These cells display antigen fragment to class II MHC molecule on the cell membrane. T cell reacts with the antigen – class II MHC molecule complex on their surface and produces co-stimulatory signals which leads to the release of cytokines and other immune mediators. Non-professional APCs are cells which do not express on MHC 2. They are stimulated by cytokines such as INF gamma. Eg-thymic epithelial cells, thyroid epithelial cells, glial cells,pancreatic beta cells, endothelial cells. Phagocytic cells are cells that protect the body by ingesting harmful foreign particles, bacteria and dead or dying cells by a process known as Phagocytosis (Fig. 1.38.24). Professional phagocytic cells include neutrophils, monocytes, macrophages, mast cells and dendritic cells. Monocytes reach an organ or a tissue from the bloodstream and they will differentiate into macrophages. Non-professional phagocytic cells include epithelial cells, endothelial cells, fibroblasts and mesenchymal cells. The fibroblasts, for example, which can phagocyte collagen in the process of remolding scars will also make some attempt to ingest foreign particle. Few other immune cells are described in Table 1.38.11. Cytokines are chemical mediators produced by cells of the immune system. Different cytokines include chemokines, interferons, interleukins, TNF and lymphokines. Innate/natural immunity, a type of non-specific immunity which is inborn. Microbial infection is detected through the pattern recognition receptors (e.g. Toll-like receptors) which survey the body for detecting infections. Adaptive/acquired immunity develops throughout our lives when we’re exposed to diseases or immunized with vaccines. Memory response is fast and highly specific. Adaptive immunity is said to be Active when it develops on contact with disease (natural) or live attenuated vaccine (artificial). Adaptive immunity is said to be Passive when it is obtained from external source. It lasts for a short time. For example, antibodies in a mother’s breast milk and immune serum (passive). There are two types of adaptive immunity, cell-mediated and antibody-mediated immunity. Cell-mediated immunity is the reaction of T lymphocytes, designed to combat cell-associated microbes. When the microbe is virus it is presented to CD8 cell and then it binds to MHC-I and further steps activated and cytokines are released. Humoral immunity is the type of immunity in which CD4+ helper T cells help B cells to produce antibodies and activate macrophages to destroy ingested microbes. Each plasma cell is derived from an antigen-stimulated B cell and secretes antibodies. This process, called affinity maturation, improves the quality of the humoral immune response. IgA is secreted from mucosal epithelia and plays a role in mucosal immunity. IgE and eosinophils provides immunity against parasites. Each microbes infect a person by different mechanism, therefore each is handled by a different part of the immune system (Fig. 1.38.25). When a bacteria enters tissue, it is readily attacked by cells of innate immunity during primary infection. These cells phagocytose and also secrete cytokines which recruit more neutrophils and macrophages to the site of infection. In addition, APC also captures antigen present it to the naïve T cells in lymph node which mount adaptive immune response. The activated T helper cells produce cytokines and promotes differentiation of B cells to plasma cells and antibody secretion. These antibodies attach to bacteria and send signals to complement proteins and phagocytic cells to destroy the microbes. All viruses, few types of bacteria and parasites, must enter cells for their survival, hence require a different immune mechanism. Infected cells with the help of MHC molecules expose the microbes on the cell’s surface, flagging down cytotoxic T lymphocytes to destroy the infected cell. Antibodies also can assist in the immune response, by destroying viruses before they enter the cell. Long-term humoral immunity is maintained by the formation of high-affinity class-switched memory B cells and long-lived antibody-secreting plasma cells. Humoral immunity promote killing of microorganism by various methods such as neutralization of antibodies, opsonization (antibody-mediated phagocytosis) and complement activation (Fig. 1.38.26). The complement pathway on activation creates membrane attack complex that targets and damages the infected cell.
1.38: Basic medical sciences for radiologists
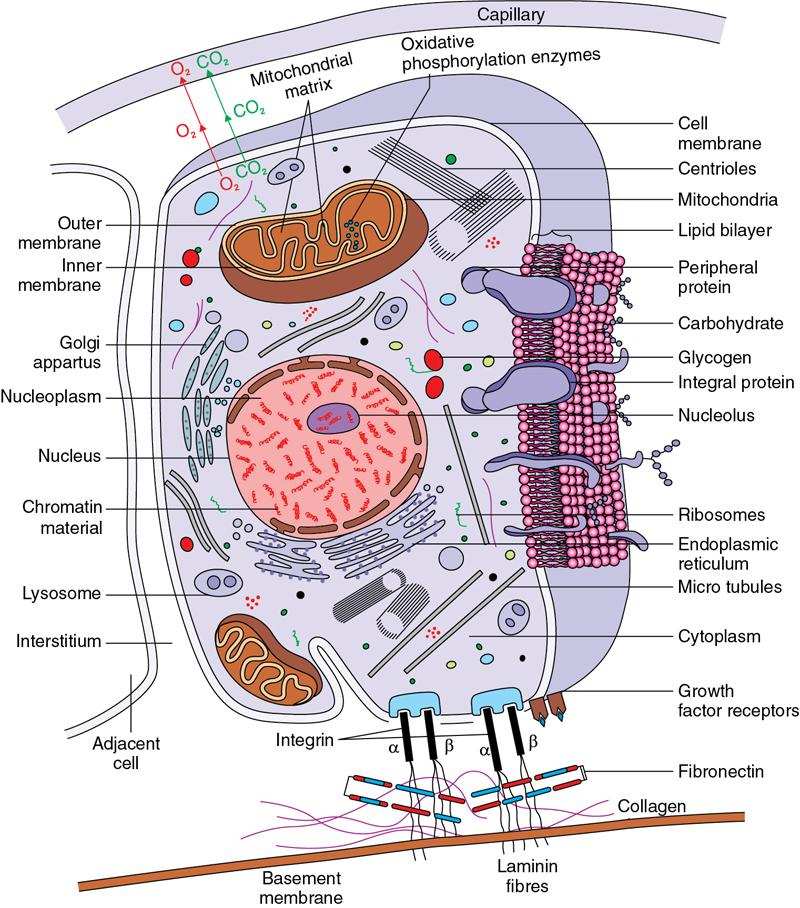
Cell and its functions
S. No
Cell Organelle
Function
1
Rough endoplasmic reticulum
Protein synthesis
2
Smooth endoplasmic reticulum
Synthesis of lipid substances, contains many enzymes involved in various cellular reactions
3
Golgi apparatus
Plays a part in protein synthesis by packaging proteins into vesicles prior to secretion
4
Cytoplasm
Glycolysis, gluconeogenesis, part of urea cycle in liver cell, pentose phosphate pathway, role in protein biosynthesis
5
Mitochondria
Kreb’s cycle, electron transport chain and produce ATP, fatty acid beta oxidation, part of urea cycle in liver cell, steroid synthesis, part of heme synthesis, storage of calcium ions, regulation of membrane potential
6
Peroxisomes
Beta oxidation of very long chain fatty acids, biosynthesis of plasmalogens, conjugation of cholic acid for bile synthesis
7
Lysosomes
Contains various enzymes involved in breakdown of proteins, lipids and carbohydrates, plays important role in autophagy
8
Nucleus
Regulates and controls gene expression, mediates replication of DNA during cell cycle, site for genetic transcription
9
Microtubules
Act as a cytoskeleton, participates in cell division, helps in cell movement, helps movement of organelles within cells
10
Nucleolus
Plays an important role in protein synthesis
Carcinogenesis
Type of Cell
Ability to Multiply
Cell Cycle
Examples
Labile cells
Continuously multiply throughout life
Short or absent G0 phase
Squamous epithelium of skin
Stable cells
They multiply only when an external stimulus is applied
They are in G0 but can re-enter the cell cycle
Liver, proximal tubules of kidney, endocrine glands
Permanent cells
They do not have the ability to multiply post-natally
They are permanently in G0
Neurons, heart muscle
Hallmarks of cancer cells
Morphological changes in cancer cells
Cellular metabolism
Carbohydrate metabolism
Mitochondrial pyruvate transporter 1 and 2 defect
Glioma
Pyruvate kinase deficiency, glucose 6 phosphate dehydrogenase defect
Hemolysis
Pyruvate dehydrogenase deficiency
Primary lactic acidosis
Fumarase enzyme mutation
Leiomas of smooth muscle and kidney
Succinate dehydrogenase mutation
Pheochromocytoma
Isocitrate dehydrogenase mutation
Glial cell tumour
NARP (Neuropathy, ataxia, retinitis Pigmentosa and Ptosis)
Mitochondrial DNA defect, ATPase6
MERRF (Myoclonic epilepsy, short stature, hearing loss, lactic acidosis, exercise intolerance)
Mitochondrial DNA defect
Leigh syndrome (subacute sclerosing encephalopathy)
Autosomal recessive, mitochondrial DNA defect/X linked, complex 1
Leber hereditary optic neuropathy
Mitochondrial DNA defect, complex 1
MELAS (mitochondrial encephalopathy with lactic acidosis and stroke like episodes)
Mitochondrial DNA defect
Pearson syndrome (sideroblastic anaemia, pancytopenia, exocrine pancreatic insufficiency, and renal tubulopathy)
Mitochondrial DNA defect
MNGIE (mitochondrial neurogastrointestinal encephalopathy)
Autosomal recessive, mitochondrial DNA defect
Kearns Sayre syndrome
Mitochondrial DNA defect/may occur sporadically
MIDD (maternally inherited diabetes and deafness)
Mitochondrial DNA defect
Mitochondrial deletion syndrome
POLG gene defect
Lipid metabolism
Peroxisome
Amino acid metabolism
S. No
Amino Acid
Function
Disorders Associated
1
Phenylalanine
Tyrosine synthesis, catecholamine synthesis, melanin synthesis, thyroid hormone synthesis
2
Threonine
Glycine synthesis
3
Glycine
4
Serine
Synthesis of glycine, cysteine, phosphatidyl serine, sphingosine, selenocysteine and choline
5
Methionine
6
Tryptophan
Synthesis of serotonin, melatonin and niacin
7
Histidine
Synthesis of histamine, glutamate, anserine, carnosine and homocarnosine
8
Arginine
Synthesis of nitric oxide, urea, creatine, ornithine and agmatine
9
Lysine
Synthesis of histones, carnitine and cadaverine
10
Glutamine
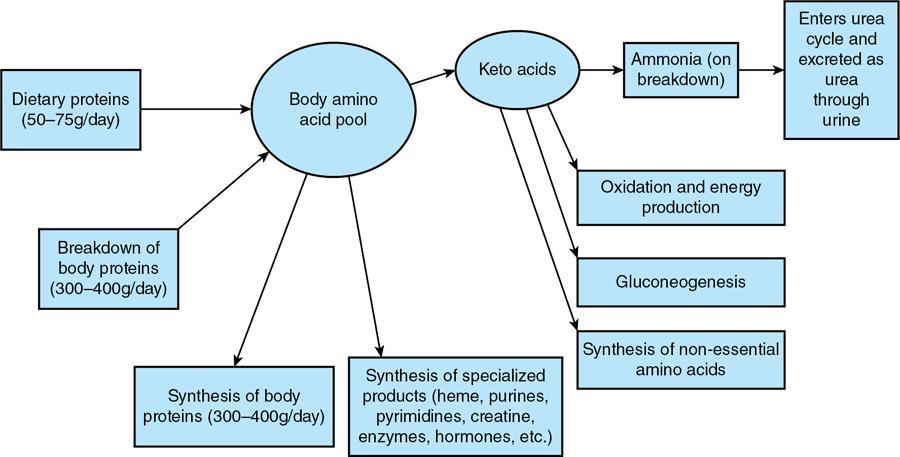
Source of ammonia, carrier of amino group from most organs including brain, purines and pyrimidine synthesis
11
Branched chain amino acids (valine, leucine, isoleucine)
All are essential amino acids
Metabolic pathways of some important amino acids
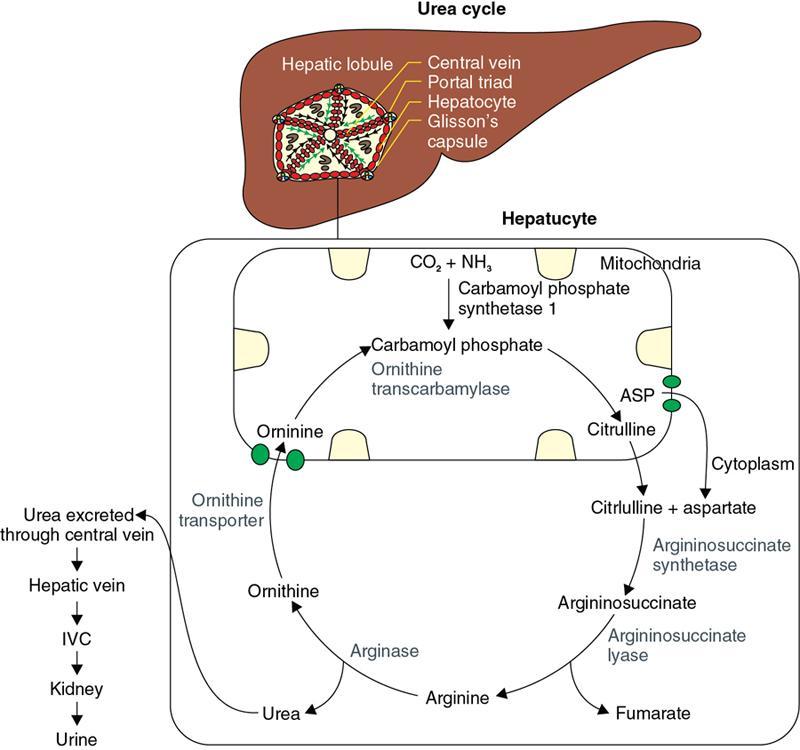
S. No
Enzyme Defect
Disorder
Biochemical Abnormalities
1
Carbamoyl phosphate synthetase 1
Hyperammonemia type 1
Hyperammonemia, greatly increased glutamine in blood
2
Ornithine transcarbamylase
Hyperammonemia type 2
Hyperammonemia, orotic aciduria, increased pyrimidine synthesis
3
Arginosuccinate synthetase
Citrullinemia type 1
Hyperammonemia, citrullinemia
4
Citrin transporter defect
Citrullinemia type 2
Hyperammonemia, citrullinemia
5
Arginosuccinate lyase
Arginosuccinic aciduria
Hyperammonemia, arginosuccinic aciduria
6
Arginase
Arginemia
Hyperammonemia, arginemia
7
Ornithine transporter defect
HHH syndrome
Hyperammonemia, hyperornithinemia, homocitrullinemia
Genetics
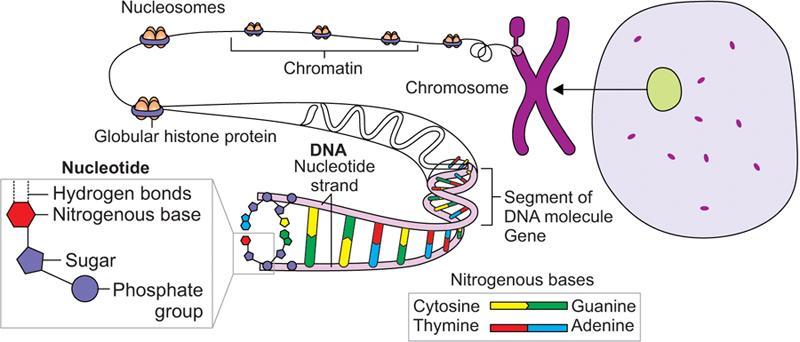
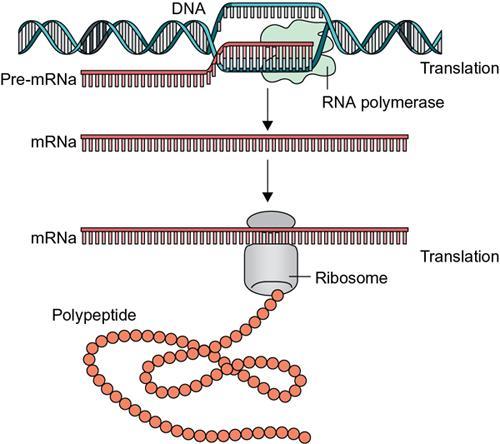
Type of RNA
Content in Cell
Location in Cell
Function
Transfer RNA (tRNA)
15%
Cytoplasm
Transfers amino acids to ribosomes for protein synthesis.
Ribosomal RNA (rRNA)
80%
Cytoplasm and rough endoplasmic reticulum
Provides structural framework for ribosomes
Messenger RNA (mRNA)
5%
Nucleus and cytoplasm
Transfers genetic material from genes to ribosomes for protein synthesis
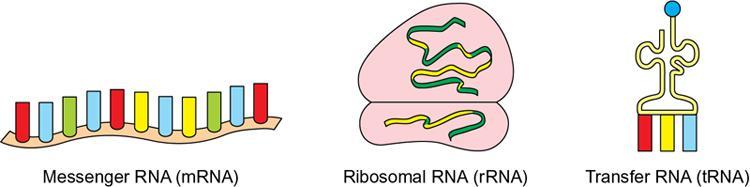
RNA
DNA
Mainly seen inside the cytoplasm
Mostly inside the nucleus
Usually 100–5000 base pairs
Millions of base pairs
Sugar is ribose
Sugar is deoxyribose
Uses uracil
Uses thymine
Guanine and cytosine content are not equal. Uracil and adenine content are not equal
Guanine and cytosine content are equal. Uracil and adenine content are equal
Easily destroyed by alkali
Alkali resistant
Genes in disease
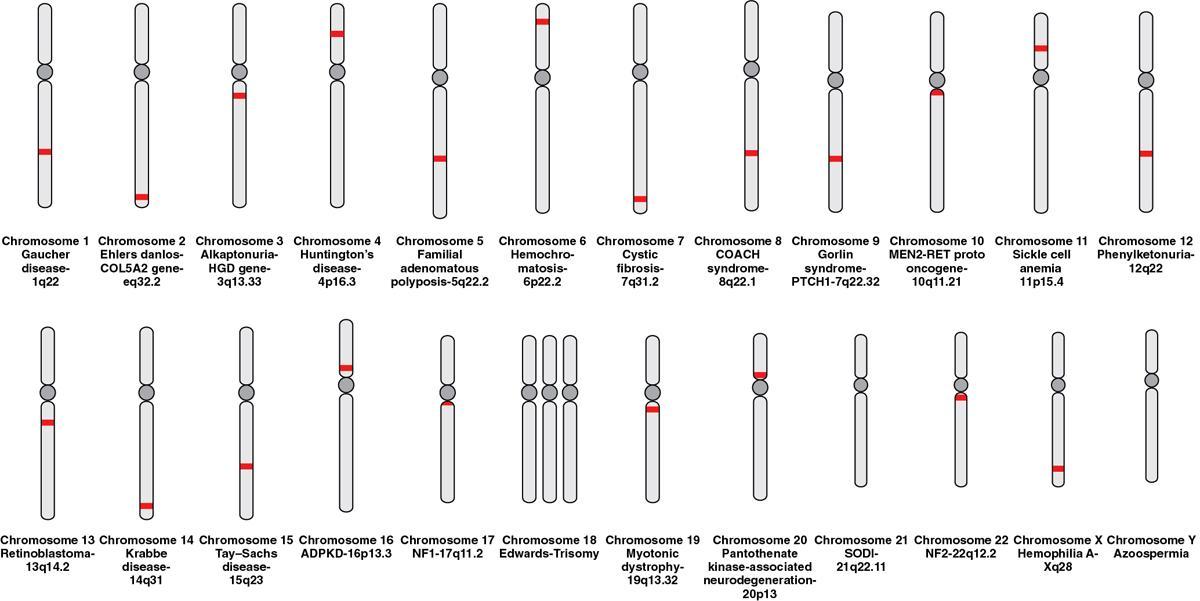
Gene Defect
Diseases Caused
PROTO ONCOGENES
EGFR (tyrosine kinase)
Lung cancer
HER2 (tyrosine kinase)
Breast cancer
H-Ras, K-Ras, N-Ras
Pancreatic cancer
c-Myc
Burkitt lymphoma
WNT (cell signalling pathway)
Medulloblastoma
TUMOUR SUPPRESSOR GENES
PTEN (phosphatase)
Breast, colon cancer
APC, GP43/Merlin (cell–cell adhesion)
NF2, colon cancer
BRCA1 (DNA repair/cell cycle checkpoints)
Breast and ovarian cancer
Neurofibromin 1 (G protein inhibitor)
NF-1
VHL (ubiquitin ligase)
Renal cell cancer
Blood
Disorders of Platelet
Disorders of Coagulation
Excessive Fibrinolysis
Vascular Defects
Quantitative – thrombocytopenia; qualitative-platelet function defect
Deficiency of coagulation/anticoagulation factors
DIC, primary fibrinolysis
Hypersensitivity vasculitis amyloidosis, Osler–Weber–Rendu disease, Ehlers–Danlos disease
Immunology
Cells and mediators involved in immunity
Types of immunity
Immune mechanism
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


