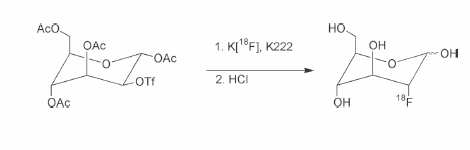
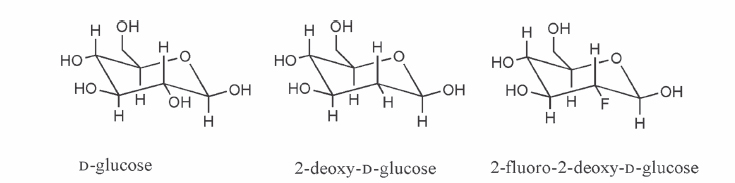
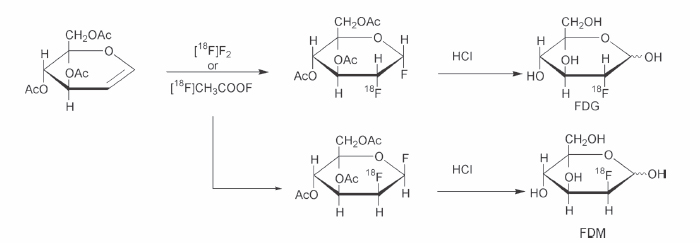
2 Without question, [fluorine 18]fluorodeoxyglucose ([18F]FDG) is the most commonly used positron emission tomography (PET) radiotracer and has demonstrated utility in a variety of applications, including neuroscience, cardiology, and oncology.1–4 A brief discussion regarding the historical development of [18F]FDG will be presented, but more detailed discussions can be found in several other recent publications.5,6 In general, there are several chemistry concerns inherent in the production of PET radiotracers. A primary concern is the impact of the relatively short half-lives of the radionuclides on the synthetic process. As a matter of practical necessity, PET radiosyntheses are designed so that the radionuclide is introduced as close to the end of the synthesis as possible. Although it is clearly advantageous to have the incorporation of the radionuclide occur as the final step in the synthetic method, this is sometimes not feasible (as is the case in the production of [18F]FDG). In most circumstances, the radio-pharmaceutical for injection must be isolated from the crude reaction mixture using a variety of methods, including solid-phase extraction methods, high-performance liquid chromatography (HPLC) methods, or some combination of the two methods. The design of 2-fluoro-2-deoxy-D-glucose was predicated upon the utility of [14C]-2-DG, a derivative of glucose where a hydrogen atom replaces the 2C hydroxyl group (Fig. 2.1). In many ways, 2-DG (and by extension FDG) and glucose are very similar. Both compounds are transported from the plasma via facilitated transport, and both compounds serve as substrates for phosphorylation by the enzyme hexokinase. However, the next enzyme in the metabolic route for these sugars, phosphohexose isomerase, requires the presence of a hydroxyl group on 2C. Consequently, 2-DG and in a similar fashion FDG are trapped in the cell as their respective 6-phosphate derivatives that are not substrates for either phosphohexose isomerase or glucose-6-phosphate dehydrogenase. This metabolic trapping leads to the utility of [18F]FDG in imaging and will be discussed further in the brief section concerning rate constants and pharmacokinetics. The demonstration that 2-FDG, in its unlabeled form, was a reasonable substrate for hexokinase and that the potential alternative substitution positions (i.e., 3F and 4F-deoxy-D-glucose, respectively) possessed significantly lower affinities for hexokinase7,8 further supported the choice of FDG as a reasonable model compound for the development of an in vivo imaging agent. Fig. 2.1 Chemical structures of glucose analogues. The first radiosynthesis of [18F]FDG took place in 1976 as the result of a long collaboration between investigators at the National Institutes of Health, the University of Pennsylvania, and Brookhaven National Laboratory.6 The first radiosynthesis of [18F]FDG was based upon the availability of [18F]F2 via the deuterium bombardment of a nickel target loaded with neon 20 [20Ne(d,α)18F, indicating the collision of an accelerated deuteron (d) with the stable nuclide neon 20 to produce an α particle (α) and the fluorine 18 radionuclide]. The reaction of [18F]F2 with a protected D-glucal (3,4,6-tri-O-acetyl-D-glucal) yielded a mixture of mannose and glucose isomers that were separable by preparative gas chromatographic methods. Acidic hydrolysis of the glucose derivative yielded [18F]FDG in amounts sufficient for human studies (Fig. 2.2).9 A variety of different approaches for the optimization of an electrophilic route to [18F]FDG were investigated.10–12 The use of [18F]acetyl hypofluorite ([18F]CH3CO2F) became one of the methods of choice. However, it was eventually demonstrated that the use of [18F]acetyl hypofluorite yielded varying amounts of the undesired isomer (2-deoxy-2[18F]fluoro-D-mannose), depending on the reaction conditions.13 One drawback of the electrophilic method (either [18F]F2 or [18F]CH3CO2F) is that only half of the label is available for incorporation into the target molecule. Another drawback is the use of carrier fluorine gas in the production that leads to reductions in specific activity. A third drawback, especially in light of the increasing utilization of [18F]FDG, was the yield limitation of the production of fluorine 18 from the 20Ne(d,α)18F reaction. The cross-sectional energies for particles in the 10 to 18 MeV range for this reaction are ~60 to 90 mCi/uA. This is significantly lower than the corresponding cross section for the 18O(p,n)18F reaction (150 to 260 mCi/uA).14 Interest in a nucleophilic radio-labeling route to [18F]FDG increased with the development of [18O] water targets capable of producing [18F]fluoride in high yield and with high specific activity. The most common method to produce nucleophilic [18F]fluoride is the 18O(p,n)18F reaction (indicating the collision of an accelerated proton (p) with the stable nuclide oxygen 18 to produce a neutron (n) and the fluorine 18 radionuclide). The oxygen 18 target material most commonly consists of enriched [18O]water.15,16 Irradiation of enriched [18O]water is capable of producing multicurie (> 70 GBq) quantities of [18F]fluoride with high specific activity in relatively short irradiation times depending on target load volumes and beam geometry. In addition, there are now methods for the separation and recovery of the enriched target material from the [18F]fluoride.17–19 Fig. 2.2 Electrophilic radiosynthetic scheme for [fluorine 18]fluorodeoxyglucose ([18F]FDG). The potential for higher [18F]fluoride yields prompted significant efforts aimed at the development of a reliable, high-yielding nucleophilic route to [18F]FDG. In most instances [18F]FDG is synthesized using an adaptation of the Julich method (Fig. 2.3).20 In the original application of this method, aqueous [18F]fluoride is added to a solution consisting of Kryptofix 2.2.2 (Merck-Schuhardt OHG, Hohenbrunn, Germany) and potassium carbonate dissolved in aqueous acetonitrile. The residual water is removed by azeotropic distillation using anhydrous acetonitrile and a stream of inert gas such as nitrogen or argon. A relatively small amount of precursor (~10 to 20 mg of 1,3,4,6-tetra-O-acetyl-2-O-trifluoromethanesulfonyl-β-D-mannopyranose) dissolved in anhydrous acetonitrile is added to the dried [18F]fluoride. The reaction mixture is heated to reflux for several minutes. Ethyl ether is used to transfer the reaction solution after cooling across a Sep-Pak silica cartridge (Waters Corporation, Milford, MA) into a second reaction vessel. This preliminary purification removes the unreacted [18F]fluoride and the Kryptofix 2.2.2. The solvents are removed, and aqueous hydrochloric acid is added to the intermediate product, 2-deoxy-2-[18F]fluoro-1,3,4,6-tetra-O-acetyl-β-D-glucopyranose. The aqueous acid solution is heated to reflux for a short period of time and then purified by passage across an ion-retardation resin followed by an alumina-N Sep-Pak and a C-18 Sep-Pak. Several aliquots of water are subsequently used to transfer all the product material from the hydrolysis vessel across the purification columns. This methodology leads to the presence of D-mannose, D-glucose, and 2-chloro-2-deoxy-D-glucose as chemical impurities in the radiosynthesis of FDG.21 This reaction scheme has been used as the basis of a computer-controlled automated synthesizer22 for the routine production of [18F]FDG (CTI, Knoxville, TN, now part of Siemens). Further modifications of this methodology have led to the development of “one-pot” syntheses for the production of [18F]FDG. These modifications include the substitution of tetramethylammonium carbonate for Kryptofix 2.2.2/potassium carbonate as the phase-transfer reagent and subsequent elimination of the silica Sep-Pak purification step. Because of these modifications, the acidic hydrolysis was performed in the same reaction vessel.23 A similar one-pot modification was reported that retained Kryptofix 2.2.2 as the phase-transfer reagent. This method also eliminated the intermediate silica Sep-Pak purification step, as well as adding a cation exchange resin to the purification column (to remove the unwanted Kryptofix 2.2.2) and an alumina N Sep-Pak to prevent fluoride ion breakthrough.24 The toxicity concerns associated with Kryptofix 2.2.2 (LD50 in rats of 35 mg/kg) have prompted the use of other phase-transfer agents such as tetrabutylammonium hydroxide or tetrabutylammonium bicarbonate. This modification has been incorporated into a commercially available synthesizer produced by Nuclear Interface. The Nuclear Interface synthesis module is flexible in that it can be set up to use either tetrabutylammonium bicarbonate or Kryptofix 2.2.2 as the phase-transfer reagent. In addition, the module can perform the hydrolysis of the radiolabeled intermediate, 2-deoxy-2-[18F]fluoro-1,3,4,6-tetra-O-acetyl-β-D-glucopyrranose, under either acidic or basic conditions. There are several other variations of this radiolabeling scheme. One variation is the use of an immobilized quaternary 4-aminopyridinium resin material for the isolation of the [18F]fluoride and subsequent incorporation into the [18F]-radiolabeled intermediate. In this process the [18F]fluoride solution is passed across the resin column, where [18F]fluoride is trapped and the bulk of the 18O-enriched water is recovered downstream. The resin-bound [18F]fluoride is dried by passing anhydrous acetonitrile across the resin column while heating the column. A solution of the precursor in anhydrous acetonitrile is then passed over the heated resin column in either a slow single pass or a reciprocating flow across the resin column. The solution containing the radiolabeled intermediate is then transferred to a hydrolysis vessel, where the acetonitrile is removed. Following the acid hydrolysis, the [18F]FDG is purified in an analogous manner to the original method described above.25 This methodology formed the basis of one commercially available synthesis unit (PET-trace FDG MicroLab, GE Medical Systems, Uppsala, Sweden). This unit uses a disposable cassette system for the reaction column, as well as transfer and addition lines that facilitate the set-up of the unit.
Basics of Fluorodeoxyglucose
Radiochemistry and Biology
Neale S. Mason and Eugene C. Lin
 Synthesis of [18F]FDG
Synthesis of [18F]FDG
Initial Method of Synthesis of [18F]FDG
Later Method of Synthesis of [18F]FDG
Electrophilic Route
Higher Yield Nucleophilic Route
Radiology Key
Fastest Radiology Insight Engine