Asif Saifuddin
Bone Marrow Disorders
Miscellaneous
Chapters 71 and 72 deal with a variety of blood-related disorders that have a major influence on imaging of the skeletal system.
Disorders of Red Cells
In late fetal life and infancy, the entire bone marrow is utilised for red blood cell (RBC) formation, supplemented by extramedullary erythropoiesis in the liver and spleen. As the child becomes older and RBC life span increases, erythropoiesis is withdrawn from the liver and spleen, then gradually from the diaphyses of the long bones, so that by the age of 25 years, active bone marrow is confined to the axial skeleton, the flat bones and the proximal ends of the femora and humeri.1 This process of withdrawal will not occur with a need for extra erythropoiesis and reverses in the presence of increased RBC destruction.
The Anaemias
Only chronic anaemias affect the radiological appearances of bone. Anaemias that do not produce reactive erythropoiesis, such as aplastic anaemia, do not affect the skeletal radiograph, but may manifest on MRI as a generalised increase in fatty marrow signal intensity (SI).1 Also, the myelodysplastic syndromes may manifest as diffuse reduction of T1-weighted (T1W) marrow SI due to marrow reconversion.1 A variety of inherited syndromes, such as Fanconi’s anaemia, may be associated with skeletal dysplasia, but these are not an effect of the anaemia per se and will not be discussed.
Chronic Haemolytic Anaemias
The Haemoglobinopathies1
The haemoglobin molecule consists of a protein (globin) and four haem groups, each with four pyrrole rings surrounding an iron atom. The protein moiety consists of 574 amino acids arranged in four spiral polypeptide chains. The different chains are designated by letters of the Greek alphabet (α, β) and the three normal haemoglobins (Hbs) A, A2 and F each contain two α chains, differing only in their second pairs.
Three types of haemoglobinopathy are found:
1. Thalassaemia: an inherited defect of HbA synthesis with inadequate manufacture of α- or β-chains.
3. Combination of thalassaemia and abnormal haemoglobin, e.g. HbS–thalassaemia.
Thalassaemia1–3
Thalassaemia is an inherited disorder and exists in two forms, the homozygous (thalassaemia major) and heterozygous (thalassaemia intermedia and thalassaemia minor), and may affect production of either α- or β-chains, resulting in α-thalassaemia or β-thalassaemia, respectively.
β-Thalassaemia is prevalent in the Mediterranean countries (Greece, southern Italy and the Mediterranean islands), while α-thalassaemia is encountered in those of West African descent.
Clinical Features.
Thalassaemia major causes severe childhood anaemia with hepatosplenomegaly, extramedullary erythropoiesis and secondary skeletal deformity. Treatment is by regular blood transfusion in infancy to maintain a haemoglobin level of 9–10 g/dL, iron chelation with agents such as desferrioxamine (DFX) to prevent/reduce iron overload, and bisphosphonates to treat the associated osteoporosis. With the introduction of successful transfusion regimes, the skeletal changes due to the disorder itself are now less commonly encountered, but osseous complications related to repeated transfusion and the effects of DFX therapy must be recognised.
Radiological Features1,2
Untreated Thalassaemia Major.
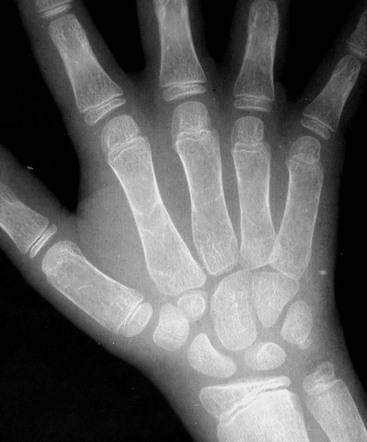
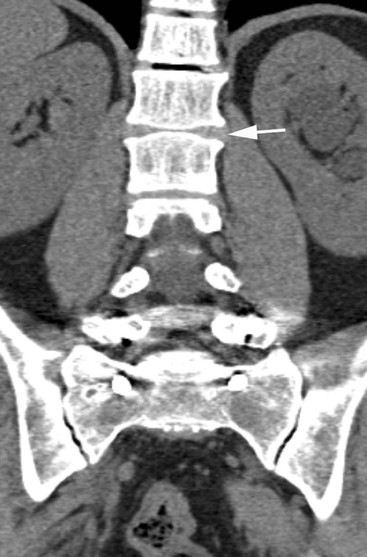
Skeletal changes in untreated children are essentially the result of chronic anaemia and marrow hyperplasia (15–30 times normal) and are most commonly seen after the age of 1 year, affecting almost all regions of the skeleton. Medullary hyperplasia results in bony expansion and cortical thinning, which in long bones produces the characteristic Erlenmeyer flask appearance, also found in conditions such as Gaucher’s disease. Within the medulla, trabecular thinning initially occurs, followed by trabecular coarsening due to new bone formation, the changes being most marked in the metacarpals and phalanges, which become cylindrical or even biconvex (Fig. 72-1). Trabecular coarsening may also be seen in the pelvis and vertebrae (Fig. 72-2). The ribs are similarly affected with club-like anterior ends, while a ‘rib-within-a-rib’ appearance also occurs due to subperiosteal extension of haematopoietic tissue through the rib cortex.
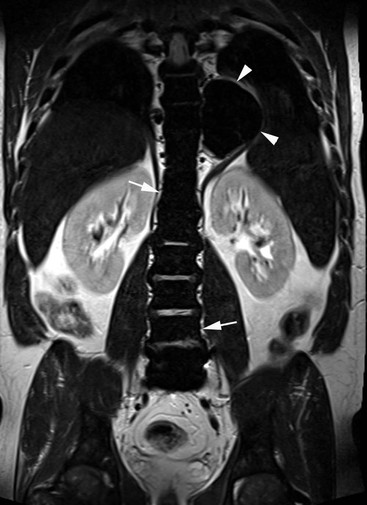
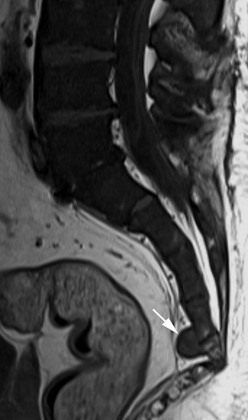
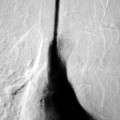
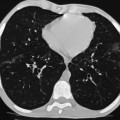
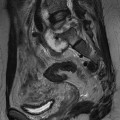
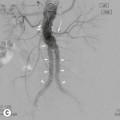
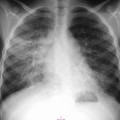
Extramedullary erythropoiesis occurs in severe cases surviving to adulthood, the most common sites being the thoracic paravertebral region by extension from the adjacent ribs, the mediastinum (Fig. 72-3) and pre-sacral region (Fig. 72-4). Epidural extension from paraspinal extramedullary erythropoiesis may result in spinal cord compression. These features are optimally demonstrated by MRI, which also shows diffuse reduction in marrow signal intensity (SI) on T1W images caused by marrow reconversion (Fig. 72-4).
With severe childhood disease, the paranasal sinuses develop poorly and often contain red marrow, accounting for the facial abnormalities (seen in 17%) and dental malocclusion. The ethmoidal cells are spared since they contain no red marrow. The diploë of the skull vault are widened, except in the occiput. These changes occur earliest and most severely in the frontal bone, producing the classical ‘hair-on-end’ appearance (Fig. 72-5). Occasionally, well-defined lytic lesions may be seen in the skull.
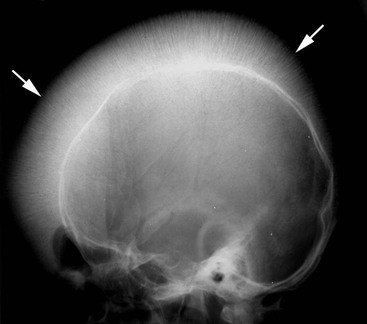
Spinal changes consist of generalised osteopenia, resulting in compression fractures and biconcavity of the vertebral bodies. Despite optimised therapy, osteoporosis may still be seen in as many as 90% of cases based on DEXA studies. Scoliosis is reported in 20% of children with thalassaemia major, while early disc degeneration is also a feature (Fig. 72-2), optimally demonstrated by MRI.
Premature fusion of the growth plates may contribute to short stature. These radiological changes are reported in ~15% of patients, generally occurring after 10 years of age and most commonly affecting the proximal humerus and distal femur. Growth arrest lines may also be seen.
Hypertransfusion.
Repeated transfusion therapy may produce iron overload and hyperuricaemia. Raised blood iron levels result in synovial and articular cartilage abnormalities, manifesting radiologically as symmetrical loss of joint space, cystic changes, subchondral collapse and osteophytosis. Chondrocalcinosis may also be seen and the larger joints tend to be more commonly affected than in primary haemochromatosis. Occasionally, the radiographic changes of gout may be evident and there is also a predisposition to osteonecrosis and osteomyelitis. Iron deposition within bone also contributes to osteoporosis and is a further cause of reduced marrow SI on MRI, particularly on T2-weighted (T2W; Fig. 72-3) and gradient-echo sequences. MRI may be used to estimate tissue siderosis.4
DFX Therapy.
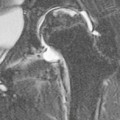
Iron chelation therapy is recognised as causing dysplastic changes in the spine and long bones, as well as growth retardation, especially when treatment is started before the age of 3 years and with higher doses. The incidence is unclear, since many cases are asymptomatic. Changes typically occur at the metaphysis/physis/epiphysis of the proximal humerus, distal femur, proximal tibia and distal radius and ulna.
Radiographs and MRI demonstrate irregularity of the metaphyseal–physeal junction with dense sclerotic metaphyseal bands, which may then extend in a ‘flame-shaped’ manner towards the diaphysis. Splaying of the metaphysis and widening of the growth plate are later features, which resemble rickets. Severe dysplasia at the proximal femur and around the knee may result in SUFE and genu varuma or valguma.
In the spine, platyspondyly and a biconvex contour to the vertebral bodies (Fig. 72-2) are seen and kyphosis may develop. DFX therapy also results in growth retardation, affecting both the axial and appendicular skeleton.
Deferiprone is an alternative treatment to DFX and has been associated with agranulocytosis and arthropathy, most commonly affecting the knees, resulting in effusion and synovitis, which can be demonstrated on MRI. Chronic changes include flattening of the femoral condyles, tibial plateau and patella.
Sickle-Cell Disease1,5,6
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


