In this article, an update is presented of the correlation of imaging and genetic findings in congenital malformations of the central nervous system (CMCNS). A nonsystematic search of the PubMed/Medline database was performed. The congenital disorders were classified in 3 groups of malformation: ventral induction disorders, cortical malformations, and congenital malformations of the posterior fossa. The highlights of genotype-imaging phenotype correlation of some congenital malformations are provided. It is hoped that developments in genotype-MR phenotype in CMCNS will foster further prognostic and pathogenic breakthroughs for the frequently associated neurologic dysfunction in children affected by these common diseases.
Key points
- •
Structural abnormalities of the central nervous system (CNS) are increasingly recognized by applying high-resolution imaging techniques, particularly MRI.
- •
As the number and complexity of recognized congenital malformations of the CNS have increased significantly, a multidisciplinary approach is mandatory, involving experts from neuroembryology, neurogenetics, neurochemistry, pediatric neurology, and pediatric neuroradiology.
- •
The MRI pattern recognition approach evolved, and integrated classifications of congenital malformations have been proposed based on embryology, genetics, and neuroimaging findings. As a result, different neuroimaging phenotypes have been observed, guiding genetic analysis and, frequently, resulting in the identification of causative genes.
- •
It is essential for every pediatric neuroradiologist to be aware of potential genotype-MR phenotype in congenital disorders of the CNS.
Introduction
Congenital malformations (CM) of the central nervous system (CNS) are commonly encountered in daily neuroimaging practice, and the significant and continuous development of the various imaging techniques, particularly MRI, has revolutionized the analysis and understanding of these disorders. Because the number and complexity of recognized CMCNS have increased significantly, a multidisciplinary approach is mandatory, involving experts from neuroembryology, neurogenetics, neurochemistry, pediatric neurology, and pediatric neuroradiology.
The MRI pattern recognition approach evolved, and integrated classifications of congenital malformations have been proposed based on embryology, genetics, and neuroimaging findings. As a result, different neuroimaging phenotypes have been observed guiding genetic analysis and, frequently, resulting in the identification of causative genes. In this context, it is essential for every pediatric neuroradiologist to be aware of potential genotype-MR phenotype in congenital disorders of the CNS.
Understanding the embryology of the CNS is crucial for every neuroscientist. Although an extensive review about this topic is not presented in this article, some concepts are important to discuss. The CNS appears in the middle of the third week of development as a thickened area of the embryonic ectoderm, the neural plate. At the onset of gastrulation, there is induction of the neural plate, which folds into the neural tube in a process called neurulation. The fusion of the neural tube occurs in a zippering process at 5 different sites in humans. By neurulation, the CNS is subdivided into its major transverse parts: prosencephalon, mesencephalon, rhombencephalon, and spinal cord. Modern neuroembryology integrates descriptive morphogenesis about these parts of the CNS with more recent insights into molecular genetic programming and data enabled by cell-specific tissue markers.
In this article, the genotype-MR phenotype correlation of the most common or clinically relevant CMCNS in the pediatric population is reviewed. The data focus on the most commonly diagnosed diseases and those in which the neuroimaging abnormalities are better defined. The PubMed/Medline database was searched using a combination of key words such as “genotype-phenotype correlation,” “genotype-imaging correlation,” and “neuroimaging-genetic correlation.” Relevant studies were defined based on current knowledge. For a more comprehensive review, the congenital disorders were divided based on an embryologic pattern, in which malformations were classified in 3 groups: ventral induction disorders, cortical malformations, and congenital malformations of the posterior fossa. In this review, malformations of the dorsal induction and spinal congenital disorders are excluded.
Introduction
Congenital malformations (CM) of the central nervous system (CNS) are commonly encountered in daily neuroimaging practice, and the significant and continuous development of the various imaging techniques, particularly MRI, has revolutionized the analysis and understanding of these disorders. Because the number and complexity of recognized CMCNS have increased significantly, a multidisciplinary approach is mandatory, involving experts from neuroembryology, neurogenetics, neurochemistry, pediatric neurology, and pediatric neuroradiology.
The MRI pattern recognition approach evolved, and integrated classifications of congenital malformations have been proposed based on embryology, genetics, and neuroimaging findings. As a result, different neuroimaging phenotypes have been observed guiding genetic analysis and, frequently, resulting in the identification of causative genes. In this context, it is essential for every pediatric neuroradiologist to be aware of potential genotype-MR phenotype in congenital disorders of the CNS.
Understanding the embryology of the CNS is crucial for every neuroscientist. Although an extensive review about this topic is not presented in this article, some concepts are important to discuss. The CNS appears in the middle of the third week of development as a thickened area of the embryonic ectoderm, the neural plate. At the onset of gastrulation, there is induction of the neural plate, which folds into the neural tube in a process called neurulation. The fusion of the neural tube occurs in a zippering process at 5 different sites in humans. By neurulation, the CNS is subdivided into its major transverse parts: prosencephalon, mesencephalon, rhombencephalon, and spinal cord. Modern neuroembryology integrates descriptive morphogenesis about these parts of the CNS with more recent insights into molecular genetic programming and data enabled by cell-specific tissue markers.
In this article, the genotype-MR phenotype correlation of the most common or clinically relevant CMCNS in the pediatric population is reviewed. The data focus on the most commonly diagnosed diseases and those in which the neuroimaging abnormalities are better defined. The PubMed/Medline database was searched using a combination of key words such as “genotype-phenotype correlation,” “genotype-imaging correlation,” and “neuroimaging-genetic correlation.” Relevant studies were defined based on current knowledge. For a more comprehensive review, the congenital disorders were divided based on an embryologic pattern, in which malformations were classified in 3 groups: ventral induction disorders, cortical malformations, and congenital malformations of the posterior fossa. In this review, malformations of the dorsal induction and spinal congenital disorders are excluded.
Malformations of the ventral induction
Malformations of the ventral induction represent a defect in the rostral closure and may result in disorders of formation, cleavage and midline development of the prosencephalon. Genetically, holoprosencephaly (HPE) and commissural abnormalities (including agenesis of the corpus callosum) are the most relevant disorders.
Holoprosencephaly
HPE is the most common developmental defect of the forebrain, with an estimated prevalence of 1 in 10 to 16,000 live births and 1 in 250 human conceptions. Children with HPE have a failure of differentiation and midline cleavage of the prosencephalon. Therefore, in the more severe and prevalent form of HPE, called alobar HPE, these patients present a crescent-shaped and single ventricle, which occupies most of the volume of the skull, fusion of the hypothalamic and basal ganglia, and no commissural structures identified. The poor prognosis in the most severe forms justifies the importance of genetic counseling in affected families.
The cause of HPE is heterogeneous: teratogens, chromosomal abnormalities, and single gene mutations can be involved. Most genes that have been implicated in HPE belong to the sonic hedgehog signaling pathway. Mutation of at least 12 different loci in 11 chromosomes has been implicated in the development of familial HPE, such as SHH , GLI2 , PTCH1 , TGIF , ZIC2 , TDGF1 , and SIX3 .
Recent data suggest specific genotype-phenotype correlations in HPE. SHH mutations result in a milder disease than mutations in the other common HPE genes. Such microforms of HPE are represented by hypotelorism, solitary central maxillary incisor, and cleft lip/palate. Mutations in ZIC2 (located on 13q32 chromosome) and SIX3 (located on 2p21 chromosome), for instance, have a high prevalence in more severe HPE types (generally, alobar subtype). ZIC2 is also associated to neuronal tube defects, mainly rachischisis, and neuronal migration abnormalities. HPE-related gene mutations in the SHH and TGIF genes have been correlated with the cause of the pituitary stalk interruption syndrome and isolated pituitary hypoplasia.
Agenesis of the Corpus Callosum
Agenesis of corpus callosum (ACC) is the most common type of commissural agenesis. Recent neonatal and prenatal imaging studies suggest that ACC occurs in at least 1 in 4000 live births and up to 3% to 5% of individuals assessed for neurodevelopmental disorders by neuroimaging. Complete and partial ACC can result from genetic, infectious, vascular, or toxic causes. Recently, Edwards and colleagues described a comprehensive classification of the clinical and genetic features of syndromes associated with ACC.
Current evidence suggests that a combination of genetic mechanisms, including single gene Mendelian, single gene sporadic mutations, and complex genetics (which may have a mixture of inherited and sporadic mutations) may be involved in the cause of ACC. For approximately 40% of individuals with ACC, the cause is identifiable (<10% have chromosomal anomalies and the remaining 30% have recognizable genetic syndromes). Genetically, it can occur either isolated or in association with congenital syndromes. The callosal anomalies are an important feature described in more than 180 syndromes. In a meta-analysis, O’Driscoll and colleagues identified 12 loci with 6 or more individuals with ACC, spread over 10 chromosomes, which is considered a high penetrance according with classification of critical region. Several of these regions are associated with common deletion or duplication syndromes in humans. The 2 ACC causative gene identified from 1 of these regions are AKT3 (located on 1q43-q44 chromosome) and FOXG1 (located on 14q12 chromosome). No phenotypic association with other CNS malformations was identified in this cohort of 374 individuals.
The ACC diagnosis is based on a finding of absent callosal fibers as visualized through neuroimaging (ultrasonography, computed tomography, or magnetic resonance [MR]). ACC is typically accompanied by a characteristic dilatation of posterior lateral ventricles (colpocephaly) associated with atypical fiber bundles (Probst bundles), which run anterior to posterior just lateral to the interhemispheric fissure. Neuroimaging can also suggest a specific syndrome based on additional imaging features.
Aicardi syndrome (AS) is the most studied syndrome associated with ACC. This syndrome is an X-linked dominant disorder (loci gene is on Xp22 chromosome). AS should be considered when ACC is related to periventricular nodular or subcortical heterotopia, polymicrogyria, cerebellar hypoplasia, posterior fossa cysts, papillomas in the choroid plexus, and microphthalamia.
ACC associated with classic lissencephaly (LIS) and perisylvian pachygyria with dysgenesis of the brainstem and cerebellum has recently been described in patients with tubulin-related cortical phenotypes. MR with these findings should be a clue to the diagnosis of TUBA1A mutation.
ACC with abnormal genitalia can be associated with ARX gene mutation. ARX gene mutation is the second most common cause of X-linked mental retardation (after fragile X syndrome) and has been associated with hydrocephaly with abnormal genitalia, LIS with abnormal genitalia, Partington syndrome, X-linked infantile spasms, myoclonic epilepsy with spasticity and mental retardation, and nonspecific mental retardation.
X-linked hydrocephalus has an incidence of 1 in 30,000 male births and is characterized by intellectual disability, spastic paraplegia, adducted thumbs, and ACC, or corticospinal tract. Most cases are ascribed to loss of function mutations L1 cell adhesion molecule ( L1CAM ) gene. L1 protein plays a key role in neurite outgrowth, axonal guidance, and pathfinding during the development of the nervous system.
Additional genetic mutations can be better studied using neuroimaging features. ACC associated with anophthalmia/microphthalmia, for example, has been described in patients with Vax1/Vax2 mutations. Vax1 and Vax2 have been implicated in eye development and the closure of the choroid fissure in animal models. Neuroimaging showing ACC in a patient with a history of a motor-sensory neuropathy has been described in Andermann syndrome, an autosomal recessive disorder caused by mutations of the KCC3 gene.
Malformations of the cortical development
Microcephaly
Microcephaly can be isolated or associated with other anomalies (often termed syndromic). When present at birth, it has been termed primary microcephaly, as opposed to secondary microcephaly, which develops later. The crucial difference between these groups is that primary microcephaly is usually a static developmental anomaly, whereas secondary microcephaly indicates a progressive neurodegenerative condition. Patients born with normal to slightly small head size (≤2 standard deviations [SD] lower than the mean) and developing severe microcephaly in the first 1 to 2 years after birth form a separate group, designated postmigrational microcephaly, because brain growth seems to slow during late gestation or the early postnatal period after normal early development.
Clinically, patients have a reduction in head circumference (HC) during pregnancy (usually at 32 weeks) or at birth. Subsequent head growth is slow, and HC worsens during infancy (<–3 SD at 6 months). Nonprogressive intellectual impairment, delay in early motor milestones, speech delay, and hyperactive behavior are common.
Pathogenesis of microcephaly is heterogeneous. Genetic and environmental factors are potential causes, and these factors may act prenatally, perinatally, or postnatally to inhibit brain growth. Genetically, isolated microcephaly is mostly Mendelian autosomal dominant, recessive, or X-linked genes. Most genes known to cause primary microcephaly compromise pathways and disturb processes such as cell cycle length, spindle positioning, or DNA repair efficiency involved in neurogenesis and, in particular, the cell cycle phases of mitosis. Ten subtypes based on the 11 genes have been differentiated ( MCPH1 , WDR62 , CDK5RAP2 , CEP152 , ASPM , CENPJ , STIL , CEP63 , CEP135 , CASC5 , and PHC1 ).
Neuroimaging of patients with autosomal recessive primary microcephalies (MCPH) shows grossly normal, proportionately small-sized brain with some degree of gyral simplification and small normal brainstem and cerebellum. A simplified gyral pattern is frequently described in patients with MCPH5, caused by mutation in the ASPM gene (chromosome 1q31). MCPH2 caused by mutations of ARFGEF2 has associated periventricular nodular heterotopia.
From an imaging point of view, 2 additional diseases are important to discuss: X-linked postmigrational microcephaly associated with mutations of CASK (calcium/calmodulin-dependent serine protein kinase) and pontocerebellar hypoplasia (PCH).
Mutations of the CASK gene at Xp11.4 have recently been reported to have a wide phenotypic spectrum, ranging from a severe form in female patients (mental retardation and microcephaly with disproportionate brainstem and cerebellar hypoplasia) to a milder form in male patients with congenital nystagmus and mental retardation. MR has been shown to be useful, showing microcephaly with disproportionate cerebellar and brainstem hypoplasia. The volume of the corpus callosum is normal in patients with CASK mutations, a finding not observed in other microcephalic patients with brainstem/cerebellum hypoplasia such as PCH, PEHO syndrome (Progressive encephalopathy with Edema, Hypsarrhythmia and Optic atrophy), 5p-syndrome, trisomy of chromosome 18, and other complex chromosomal abnormalities. CASK is a protein that induces transcription of genes containing TBR1 binding sequences, such as RELN , required for normal development of the cerebrum, brainstem, and cerebellum.
PCH caused by mutations in transfer RNA splicing endonuclease subunit genes ( TSEN54 , TSEN2 , TSEN34 ) is a prenatal onset neurodegenerative disorder, in which significant microcephaly develops after birth. Recently, MR data showed that the common homozygous mutation in TSEN54 can be predicted reliably from the PCH type 2 MR phenotype, described as a dragonflylike cerebellar hemisphere and a flat pons associated clinically with dyskinesia or dystonia, neonatal irritability, central visual impairment, absence of optic atrophy, and severe cognitive and motor impairment.
Megaloencephaly
A practical approach to patients with macrocephaly divides the megaloencephaly (MEG) syndromes in 2 groups: (1) MEG with normal cortex and (2) MEG with cortical dysplasia.
Examples of genetically defined MEG syndromes with a normal cortex are Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome (BRRS), Proteus syndrome, autism spectrum disorders with macrocephaly (ASDM), and Sotos syndrome. These disorders are also named PTEN hamartoma tumor syndromes and refer to a group of clinical syndromes of aberrant growth caused by mutations in the PTEN tumor suppressor gene.
Neuroimaging of patients with Cowden syndrome can show dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease), and patients with BRRS can present with white matter cysts, complex dural arteriovenous fistula, and progressive spinal epidural lipomatosis. Brain abnormalities are not common in Proteus syndrome; when present, hemimegalencephaly and migrational disorders are typically seen, commonly with an associated seizure disorder. Maxillary and mandibular dysmorphism may occur, including unilateral condylar hyperplasia. Subcutaneous fatty, fibrous, lymphangiomatous masses commonly seen in this syndrome may involve the neck and face, leading to disfigurement and potential airway compromise. Imaging of patients with ASDM with mutation of the PTEN gene shows a suggestive pattern of multifocal white matter lesions associated with perivascular spaces. MR studies of patient with Sotos syndromes show ventricular abnormalities, extracerebral fluid spaces, midline abnormalities and migrational abnormalities.
MEG syndromes with cortical dysplasia have been noted to arise as sporadic overgrowth disorders, and recent data from the literature led to the characterization of 2 MEG syndromes by physical and neuroimaging anomalies: megaloencephaly-capillary malformations syndromes (MCAP) and megaloencephaly-polymicrogyriapolydactyly-hydrocephalus syndrome (MPPH). Riviere and colleagues identified a striking correlation between MCAP and MPPH, with mutations in PIK3CA , PIK3R2 , and AKT3 genes. Neuroimaging of patients with MEG syndromes shows ventricular enlargement, cortical dysplasia (usually polymicrogyria), cerebellar tonsillar ectopia, thickening of the corpus callosum, and cranial asymmetry.
Cortical Dysgenesis with Abnormal Cell Proliferation but Without Neoplasia
Cortical dysgenesis with abnormal cell proliferation can be diffuse, focal, or multifocal. Focal cortical dysplasia (FCD) represents a diverse group of neuron structural abnormalities and can be classified in subtypes I to III, by histopathologic criteria. Evidence has suggested that FCD is unlikely to be associated with a single gene mutation and is probably related to polymorphism mutations in the regulatory elements or to involve multiple genes such as TSC1 and notch/Wnt signaling pathways. Regarding hemimegaloencephaly, data suggest that somatic mutations of the PI3K-AKT-mTOR pathway limited to the brain may represent a potential cause of asymmetric enlargement of the brain.
Malformations with Neuroependymal Abnormalities: Periventricular Heterotopia
Periventricular heterotopia (PVH) has been classified in 2 groups based on the location of the nodules: anterior or posterior. Genetically defined anterior PVH with an autosomal dominant inheritance has been described in patients with mutations on chromosomes 1 and 5. Mutations of filamin A ( FLNA ), a protein that crosslinks actin filaments into orthogonal networks in cortical cytoplasm, has been described in patients with an X-linked inheritance associated with bilateral PVH.
Malformations Caused by Generalized Abnormal Transmantle Migration
LIS and subcortical band heterotopia (SBH) are examples of congenital malformations related to generalized abnormal transmantle migration. Neuroimaging with anterior predominant or diffuse classic (4-layered) LIS and SBH has been shown in patients with DCX mutation at Xq22.3–q23. In addition, a frontal predominant mild LIS with severe hippocampal and cerebellum hypoplasia has been associated with RELN mutation and VLDLR mutation. For the posterior predominant or diffuse classic (4-layered) and 2-layered (without cell-sparse zone) LIS and SBH, TUBA1A mutations, Miller-Dieker syndrome (4-layered) with deletion at 17p13.3 (YWHAE telomeric to LIS1), and posterior or diffuse LIS (isolated LIS sequence, 4-layered) or posterior SBH with LIS1 deletions or mutations at 17p13.3 have been reported. Mutations in ARX at Xp22.13 have been shown in patients with X-linked LIS (3-layered, without cell-sparse zone) with callosal agenesis and ambiguous genitalia (XLAG).
Malformations Caused by Abnormal Terminal Migration and Defects in Pial Limiting Membrane
Cobblestone malformation (former type 2 LIS) is a severe neuronal migration disorder characterized by protrusions of neurons beyond the first cortical layer at the pial surface of the brain. It is usually associated with eye anomalies and congenital muscular dystrophy (CMD). Cobblestone malformations occur in a graded series of CMDs, with brain involvement associated with reduced glycosylation of α-dystroglycan, which, from least to most severe, include CMD with mental retardation and microcephaly without obvious cortical malformation; CMD with mental retardation and isolated cerebellar hypoplasia and dysplasia; Fukuyama CMD (FCMD); muscle-eye-brain disease (MEB); and Walker-Warburg syndrome (WWS).
For the genetically defined cobblestone malformations (WWS, MEB, or FCMD) with frontal predominant abnormalities, POMT1 mutation, FKTN mutation associated with retinal abnormality (MEB-like), and LARGE mutation have been shown. Frontal predominant cobblestone has also been shown with GPR56 mutations (bilateral frontoparietal polymicrogyria [BFPP]).
For the genetically defined cobblestone malformations (WWS, MEB, or FCMD) with posterior predominant abnormalities, LAMA1A and LAMC3 mutation (lacks CMD) has been reported. Cobblestone malformations associated with congenital disorders of glycosylation (CDG) have been reported in patients with SRD5A3 mutation.
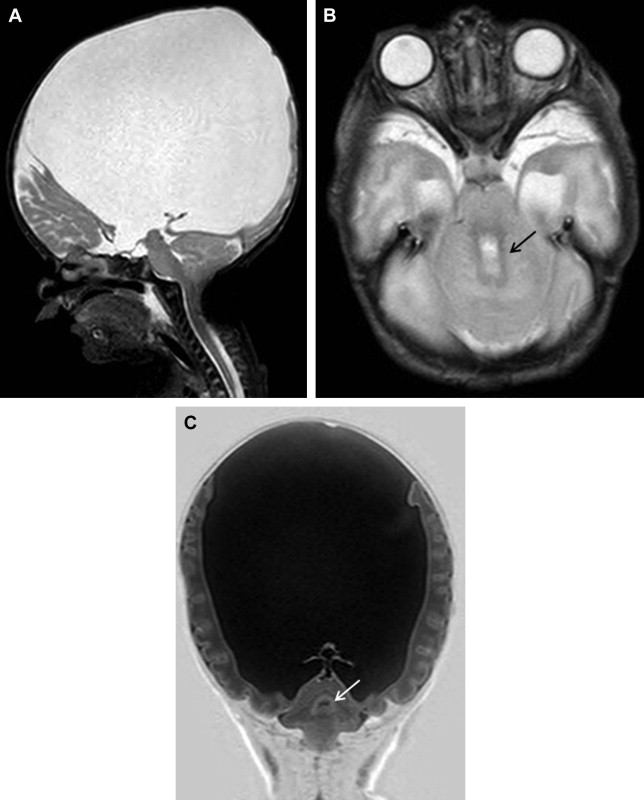
Polymicrogyria
Genetic heterogeneity is expected in polymicrogyria, because many types seem to be inherited as single gene disorders. In some cases, inheritance is multifactorial or complex, with interaction of several modifying genes, each with a small additive effect, and environmental factors ( Fig. 1 ).
Polymicrogyria, including the most common perisylvian subtype, has been associated with several chromosomal deletion and duplication syndromes, including the common deletion at 22q11.2 (DiGeorge syndrome). Multiple observations of familial polymicrogyria have been reported. Three loci of interest for the most common bilateral perisylvian form of polymicrogyria have been identified on the X chromosome, yet only 1 patient has been identified with a mutation in a gene at 1 of these loci, the SRPX2 gene at Xq22. Other recessive pedigrees show a frontoparietal distribution, with mutations of the GRP56 gene or a diffuse distribution associated with peroxisomal disorders. Mutations in the TUBB2B gene have recently been identified in 4 patients with asymmetric polymicrogyria, and functional studies suggest that this gene is required for neuronal migration.
For BFPP, previous studies have indicated a gene, GPR56 , on chromosome 16. Analysis of the GPR56 gene expression pattern suggests an essential role in regional patterning of the frontoparietal cortex. For perisylvian polymicrogyria, different patterns of inheritance, including X-linked dominant, X-linked recessive, autosomal recessive, autosomal dominant with reduced penetrance, autosomal recessive with pseudodominance, and autosomal dominant have been suggested, showing the genetic heterogeneity of BFPP. A unilateral or bilateral polymicrogyria associated with a velocardiofacial syndrome, DiGeorge syndrome, or conotruncal heart malformations has been described in patients with deletion of chromosome 22.
Midbrain and hindbrain malformations
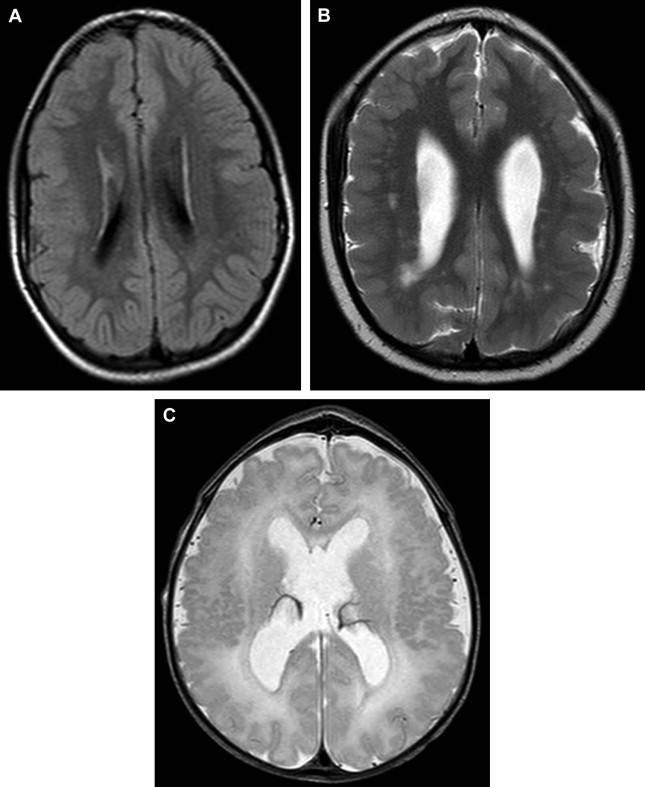
Dandy-Walker Malformation
The Dandy-Walker malformation (DWM) is a heterogeneous disorder that has been associated with several malformation syndromes and cytogenetic abnormalities ( Fig. 2 ). Over the past decade, several genetic loci have been implicated in the pathogenesis of DWM and other linked disorders, such as cerebellar vermis hypoplasia (CVH) and megacisterna magna (MCM), including the ZIC1 , ZIC4 , FOXC1 , PAX3 , NDUFA4 , and PHF14 genes.
DWM, CVH, and MCM are generally classified as different disorders; however, they share similarities in appearance, and it is not known whether they represent distinct entities or share a common pathogenesis. The low empirical recurrence rate shown in nonsyndromic DWM suggests a polygenic model for the malformation.
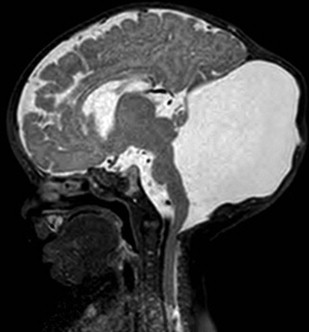
Rhombencephalosynapsis
Rhombencephalosynapsis (RES) is a congenital malformation that consists of cerebellar hemisphere continuity, usually in the dentate nuclei and superior cerebellar peduncles caused by total or partial agenesis of the vermis. The classic MR findings are fusion of the cerebellar hemispheres, with continuous white matter tract crossing midline, fourth ventricle in a diamond shape, and absence of primary fissure. It might be related to aqueductal stenosis (causing hydrocephalus) or dysgenesis of the corpus callosum, mainly the posterior portion ( Fig. 3 ).