Classification
Examples
Magnitude of risk
High penetrance
BRCA1, BRCA2, TP53, PTEN, STK11, LKB1, CDH1
25–85 % lifetime
Intermediate penetrance
CHEK2, ATM, BRIP1, BALB2
Two to threefold increased
Low penetrance
Numerous SNPs from GWAS studies
1.5-fold increased or less
The inherited predisposition to developing breast cancer is most often associated with mutations in the BRCA1 and BRCA2 genes [9]. Approximately 5–10 % of women with a family history of breast cancer will carry a mutation in one of these two genes. While damaged BRCA genes occur in most populations, the frequency of the altered alleles is higher in certain populations, such as Ashkenazi Jews. For those who do carry an altered BRCA gene, their lifetime increase in breast cancer risk ranges from 25 % to 85 %. Elevated risk of ovarian cancer is also seen in these women. The histopathology of tumors with mutant BRCA genes differs from those occurring sporadically. Tumor grade is often higher and expression of hormone receptors (estrogen receptor (ER) and progesterone receptor (PR)) is often lower than in sporadic tumors. HER2 expression is infrequent. As a result, these tumors are often classified as triple negative or basal subtype. Other high penetrance gene mutations known to increase the risk of a breast cancer diagnosis include TP53, PTEN, and ataxia telangiectasia (ATM) genes, which all increase the risk of breast cancer by eight to tenfold.
Ethnicity has been shown to influence the risk of developing breast cancer beyond known risk genes. Both black women and Asian women have lower incidence of disease [5, 12]. However, women from both groups tend to present at a younger age with more advanced disease. In the case of black women, lower usage of mammography may contribute, but a genetic component may also play a role. The presentation at a more advanced stage in Asian women may be a result of having denser breast tissue on average, which is an impediment to mammographic identification of breast tumors [12].
9.2.2.2 Environmental Factors
There has been a great deal of interest in modifiable factors that influence development of breast cancers. Although there is a large body of work demonstrating genetic basis for this increased risk from alterations in genes that increase cancer predisposition, such as BRCA1 and BRCA2, there is also ample evidence for a shared environmental component to the increased risk. These have the potential to be inexpensive and broadly applicable means to reduce the burden of disease across populations. The clearest evidence for environmental influence on risk is the change in breast cancer incidence in genetically similar populations that differs only by geographic location (see discussion above). An example comes from studies demonstrating that incidence of breast cancer is higher in urban Chinese women than in Chinese women who live in a rural environment [7]. The nature of the environmental factors that cause this difference are not yet clear.
Although high fat diets have been implicated in increased risk of breast cancer in observational studies, meta-analyses have not substantiated the adverse risk effects of dietary fat [13]. Fruits and vegetables appear to confer protective effects, while alcohol increases risk. Other nutrients, such as vitamins and beta-carotene, have been investigated as risk modifiers. No clear answer has emerged about the magnitude of effect of these nutritional factors.
Physiological factors are a clearer source of risk modification for breast cancer. Obesity has been shown to both increase breast cancer incidence and mortality. One study (Women’s Health Initiative) demonstrated a 2.5-fold increase between those with high body mass index versus those with low body mass index [14].
9.2.2.3 Hormonal Factors
The development of breast cancer is strongly influenced by endogenous hormones. Epidemiological studies have consistently shown a relationship between hormonal status and breast cancer risk. Early age at menarche, nulliparity, late first full term pregnancy, and later menopause all increase risk of developing breast cancer [15, 16].
The incidence of breast cancer increases with age; the increase in risk is steep up to menopause. Post-menopause the risk continues to increase, however, the rate of increase diminishes to approximately 15–20 % of that prior to menopause. The dramatic drop in the rate of increase implicates ovarian activity and endogenous estrogens in breast cancer etiology. The role of ovarian hormones is further substantiated by similar decreases in risk following oophorectomy. In contrast, events that increase hormone exposure, such as hormone replacement therapy for menopause symptoms, increase the risk. Early age at menarche and late age at menopause both increase the risk of developing breast cancer. Taken together, these factors indicate that extended exposure to hormones, especially estrogen, increase the risk of breast cancer [14].
The interplay between breast cancer risk and pregnancy has been closely studied. The relative risk for women who have their first full term pregnancy after 30 years of age is two to fivefold greater than for women who complete a pregnancy before 18. The risk for women who do not become pregnant is approximately 1.4-fold higher than for those who do. Interestingly, the risk of developing breast cancer increases transiently following pregnancy. This increase lasts approximately 10 years, but then is associated with a more durable protective effect. Exposure to high levels of estrogen in utero is associated with an increased risk. Breast feeding reduces risk of a breast cancer diagnosis.
As can be seen from this discussion, the interplay between age, reproductive hormones, pregnancy, and breast feeding modulates breast cancer risk substantially. It is thought that this combination of factors may play a significant role in the difference in breast cancer risk between developed and developing nations discussed earlier in this chapter.
9.3 Molecular Mechanisms
9.3.1 Introduction
Breast cancer, because of its significant negative impact on mortality and quality of life, has been a main focus in the war on cancer initiated 40 years ago. A collateral benefit of the intense research focus on breast cancer has been knowledge that is generalizable to other cancers. A great deal of the knowledge that has been generated about the underlying biology of cancer has its roots in breast cancer research.
Various systems within the cancer cell are disturbed resulting in the malignant phenotype that is observed: incessant growth, self-sufficiency in growth signaling, resistance to apoptosis, invasion and metastasis, and abnormal angiogenesis. These systems include signal transduction pathways for mitogenic signals, cell cycle control systems, DNA repair systems, and epigenetic gene expression modification systems. Some of the molecular details of these systems are reviewed in the following sections.
9.3.2 Signal Transduction
9.3.2.1 Estrogen Signaling
Observations dating back more than a century suggested the importance of estrogen and its receptor in breast biology and breast cancer. Interference with this endocrine axis affected breast morphology and, as it was later shown, breast cancer growth [17]. Observations from the 1950s suggested that the receptor for estrogen could be quantitated and that its concentration varied between breast tumors. By the 1970s it had been demonstrated that breast cancer patients could be stratified by ER concentration in their tumors with those responding to endocrine-based therapy grouped in the ER+ category [15]. These insights formed the foundation for the use of estrogen antagonists as therapeutics and measurement of estrogen receptor as a diagnostic biomarker. Both are among the earliest examples of the application of molecular medicine.
Estrogen and its receptor have manifold effects on gene expression in relevant cell types. Classically, estrogen binds to its receptor in the nucleus of the cell, which then activates the receptor for DNA binding and transcription activation via estrogen response elements (EREs) in the promoters of estrogen-responsive genes. ER can also activate gene expression independent of EREs. It does so by forming complexes with other transcription factors, including JUN and AP-1, which then stimulate transcription via their cognate binding sites in gene promoters for genes such as ovalbumin, IGF-1, and CCND1. In addition to transactivation of gene expression, estrogen receptor intersects diverse pathways involved in mitogenic signaling in a genome-independent manner. For example, evidence suggests that membrane-bound ER can mediate activation of the MAP kinase signaling pathway by estrogen in several cell types [18].
The exact effect produced by activated estrogen receptor depends not only on cellular context, but also on the chemical structure of the ligand bound by the receptor. Therefore, a great deal of effort has been expended to understand the structure-activity relationships involved with ER. The fruits of these efforts have been effective anti-estrogen therapeutics, including tamoxifen, raloxifene, anastrozole, and others. New knowledge continues to emerge about pharmacological intervention in cancer through study of ER in breast cancer.
9.3.2.2 Growth Factor Signaling
Growth factor receptors are a major target for therapeutics in cancer. Much of our understanding of how to leverage information about growth factor receptors in cancer has been built upon the foundation established by studies of HER2 as an oncogene and as a therapeutic target in IBC (Fig. 9.1).
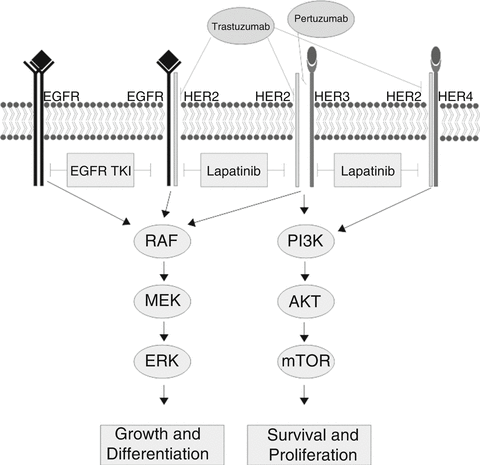
Fig. 9.1
Signaling through the HER2 pathway. The Erb-B/HER family of growth factor receptors and their signaling pathways. HER2 is shown dimerizing with other family members. Receptors and heterodimers targeted by therapeutics are indicated (Source: Eric R. Schuur)
Initial observations established that over-expression of HER2 (ERB-B2) was a marker for breast cancers that recurred early, had a more aggressive course, and were usually ER negative (ER-) [19]. No effective treatment was available for these patients—HER2+ tumors often did not respond to conventional chemotherapeutic regimens available at that time. These patients often wound up in clinical trials in an effort to find a therapy that would control their disease.
Out of translational research efforts emerged a humanized monoclonal antibody directed against the HER2 extracellular domain, trastuzumab. Although at that time there had been limited success with monoclonal antibodies as therapeutics, trials were initiated in which trastuzumab was added to standard adjuvant chemotherapy. The results of these trials clearly demonstrated that targeting HER2 with trastuzumab in women with HER2 positive disease produced improvements in disease free survival versus adjuvant chemotherapy alone [20].
As is too often the case in cancer, patients will have an excellent response to therapy, only to have that same therapy become ineffective some weeks, months, or years later. The HER2 system also exhibits this behavior. Further examination of the biology of HER2 signaling in IBC has yielded insights into the mechanisms of resistance (Fig. 9.1).
Studies have shown that HER2 must dimerize with itself or other family members to signal [21]. A form of resistance to anti-HER2 agents is based on dimerization of HER2 with family members EGFR (HER1) or HER3, allowing signal transduction in the presence of HER2 inhibitors. A second monoclonal antibody to HER2, pertuzumab, blocks dimerization and is also an effective IBC therapeutic and, importantly, can act together with trastuzumab to increase therapeutic effect [22]. This result supports the principle that blocking “rescue” signal transduction can overcome therapy resistance.
Extending our understanding of growth factor receptor signaling pathways has enabled the development of a series of other breast cancer therapeutics. Other growth factor receptors have been targeted, such as the epidermal growth factor receptor (EGFR, also known as HER1) and the insulin-like growth factor receptor 1 (IGF-1R), either with monoclonal antibodies or with small molecule drugs. Downstream components of these signaling pathways have also been successfully targeted in drug development. Drugs that target these signaling components, including imatinib and sorafinib, are being tested for activity as breast cancer therapeutics [21].
9.3.3 Cell Cycle Control
Several gene that are implicated in breast cancer oncogenesis function in regulating progression through the cell cycle, including TP53, ATM, CCND1, and CHEK2. TP53 encodes the P53 tumor suppressor protein, which is one of the most frequently mutated genes in many different types of cancer. The P53 protein is a multifunctional protein that regulates its target genes in response to various cellular stresses, including DNA damage, and thereby arrests the cell cycle or induces senescence, apoptosis, DNA repair, or metabolism alterations, depending on the context. CCND1 encodes the cyclin D1 protein, which functions to regulate progression through the cell cycle by interacting with cyclin dependent kinases 4 and 6. Cyclin D1 is itself regulated by the tumor suppressor protein, RB. The ATM gene encodes a PI3/PI4-kinase family protein that regulates cell cycle checkpoints in response to DNA damage from sources such as ultraviolet radiation. The CHEK2 gene encodes a protein that is a cell cycle checkpoint regulator and putative tumor suppressor. The protein is activated by DNA damage and cell cycle blockage and interacts with P53 and other proteins to induce cell cycle arrest in G1. Although insights into breast cancer biology have come from study of these genes, their biochemical nature has proved more difficult to target with therapeutics than the signal transduction pathways.
9.3.4 DNA Damage and Genomic Instability
9.3.4.1 DNA Damage Repair Pathways
Progressive DNA damage is a feature of most cancers and stems from loss of function of repair pathways. Damage to the double stranded DNA molecule can take several forms: single strand cleavage or base substitution, double strand breaks, or interstrand crosslinks. Because the information encoded by the DNA molecule needs to be protected, the cell deploys multiple systems to guard against this damage happening and to repair it if it does occur. Single nucleotide excision repair and strand ligation can restore single strand breaks, some of which are involved in the cancer syndrome, xeroderma pigmentosum (XPA, XPG). Other genes in this system have been implicated in non-small cell lung cancer (ERCC). A different set of enzymes is required to repair interstrand crosslinks, including some (FANCD1, FANCI, FANCJ, FANCN, PALB2) which are implicated in Fanconi anemia. Double strand breaks are repaired by yet another system of enzymes, which include the BRCA1 and BRCA2 genes responsible for many hereditary cases of breast cancer [23].
The BRCA genes both code for large multifunctional proteins that genetically behave much like classic tumor suppressor genes. In addition to influencing the risk of breast cancer, mutations in these genes increase the risk of ovarian cancer, as well. They encode proteins that have multiple functions in maintaining genomic stability by facilitating DNA double strand break repair by homologous recombination. Loss of heterozygosity of the BRCA genes results in deficient DNA repair and accumulation of additional DNA damage, which contributes to oncogenesis. In the absence of BRCA function, cells fall back on poly(adenosine phosphate-ribose) polymerase-1 (PARP1) for DNA repair. A synthetic lethal therapeutic strategy that is in clinical testing inhibits PARP activity to tip the balance in these mutant breast epithelial cells toward cell death, rather than rescue DNA repair [24].
9.3.4.2 Genomic Instability in Breast Cancer
The breakdown of the DNA damage repair systems leads to accumulating DNA damage, and, ultimately, progressive genomic instability and aneuploidy, both hallmarks of cancer. The instability is characterized by point mutations and gene amplifications, with an example of the latter being amplification of 17q12 where the HER2 gene is located. Later, deletions or insertions of DNA segments and ultimately loss or duplication of entire chromosomes occurs. The latter stages of aneuploidy are characteristic of late stage, fatal cancer.
Genome stabilizing systems that have been shown to malfunction in breast cancer include cell cycle checkpoint (TP53, CCDN1) and DNA repair (BRCA1 and BRCA2), as described above. In addition to these systems, restriction point controls for entry into the cell cycle, spindle assembly checkpoints, and cellular senescence are additional mechanisms for controlling DNA damage to the individual cell and thereby avoiding progressive genomic instability [25].
Details of processes of genome instability have consequences for cancer therapeutics. For example, traditional cytotoxic therapeutics, such as the platinum compounds, were developed empirically and induce cell killing through DNA damage. At least in some types of cancer, this cell killing requires P53; the loss of P53, with the attendant genome destabilizing effects of that loss, may, in fact, accelerate genome damage and cancer progression. In other types of cancer, these same compounds may be very effective in inducing cell death, without the need for P53 [26]. Hence, an understanding of the mechanisms of genome destabilization and the consequences of cellular context will be important to understand.
9.3.4.3 The Genomic Landscape of Breast Cancer
As noted above, most breast cancers of are sporadic and result from DNA changes that accumulate over time. In agreement with the precepts of oncogenesis, breast cancers have been shown to accumulate DNA damage resulting in oncogenic alterations, with the typical breast adenocarcinoma harboring 60–80 somatic mutations [27]. Efforts to define the range of somatic alterations and place these in clinical context are underway and have recently borne fruit [10].
A catalogue of these changes will not be sufficient to define their role in oncogenesis. The vast majority of these mutations are passenger mutations, unlikely to influence cancer phenotype [11, 27]. The driver mutations responsible for development of the disease can be challenging to uncover. The required evidence that connects these mutations in driver genes to the oncogenic process includes both association with the cancer phenotype in clinical samples, as well as experimental demonstration that the alterations can participate in oncogenesis or progress in model systems. Current high throughput systems are defining the genomic landscape of DNA alterations, identifying potential driver mutations. Often altered genes have available experimental data on oncogenesis, which can facilitate assignment as a driver gene and therefore potential therapeutic target.
The picture that is beginning to emerge is that of a genomic landscape with a few genes that are very frequently mutated in breast cancer and a much larger assortment of genes that are mutated in a small proportion of breast cancers (Table 9.2). The relative roles of the frequently mutated genes and those with lower frequency alterations is still not clear. However, recent evidence suggests that it may be possible to group the infrequently mutated genes in a smaller number of pathways or phenotypic groups, effectively reducing the complexity of the genomic landscape [29]. Examples of the frequently mutated genes in somatic samples include some of the same genes that are altered in familial predispositions: TP53 and BRCA1/2. Others examples of often mutated genes include PIK3CA, GATA3, MAP3K1, and PTEN.
Table 9.2
Frequently mutated genes in breast cancer
Gene | # cases | Percentage |
|---|---|---|
TP53 | 187 | 37 % |
PIK3CA | 180 | 36 % |
GATA3 | 54 | 11 % |
MAP3K1 | 39 | 8 % |
MLL3 | 37 | 7 % |
CDH1 | 33 | 7 % |
MAP2K4 | 21 | 4 % |
RUNX1 | 18 | 4 % |
PTEN | 17 | 3 % |
TBX3 | 13 | 3 % |
In addition to mutation, somatic alterations include altered copy number. Examples include HER2 at 17q12 (noted earlier) and cyclin D1 at 11q13. Overexpression of the latter gene also contributes to the cancer phenotype and has prognostic significance. Integration of the genomic changes in breast cancer with information on changes in RNA and protein expression and changes in epigenetic modifications will increasingly form the foundation for molecular medicine.
9.3.5 Genetic Alterations Are Reflected in RNA and Protein
The cell is often viewed as a system of networked pathways consisting of DNA, RNA, protein, and other components [30]. Disturbance in one component (e.g. DNA as above) will be reflected in alterations in the other components and other pathways that are integrated with it. DNA alterations found in IBC include all of those known to induce cancer, including point mutations, small insertions or deletions (indels), and amplification. The prevalence of DNA alterations in breast cancer is intermediate, with 60–80 alterations typically found in tumors.
A consequence of DNA damage is alteration of the RNA content of the cell, with the nature of the alteration specific to the type of DNA damage that has occurred. With the development of highly parallel methods to measure gene expression, such as DNA microarrays, it has become possible to measure the concentration of RNA molecules as a snapshot of the cell’s gene expression state. The ability to easily gather expression information on thousands of genes simultaneously and associate that information with phenotypes has enabled association of altered gene expression with cancer phenotypes. This has connected molecular information from tumors to their clinical behavior in a more universal way than previously possible.
The primary consequences of DNA damage at the protein level are alterations in expression level or altered protein structure. In the case of breast cancer the result is altered activity of several signal transduction pathways that influence cell division and angiogenesis. The classes of proteins involved include steroid receptors, receptor tyrosine kinases, intracellular kinases, and transcription factors.
9.3.6 Epigenetics in Breast Cancer
DNA is packaged into chromatin with histones and other accessory proteins in the nucleus. Epigenetic machinery serves to modulate the interaction of DNA and packaging proteins to enable transcription to occur in a selective fashion. There are two principle epigenetic processes: DNA methylation and histone acetylation. Methylation of cytosine bases in promoters functions to reduce transcription of the associated genes. Acetylation of histone proteins modulates the association of histone with specific promoters, again influencing transcription of these genes. Epigenetic processes are critical to ensure stable control of gene expression in the processes of cell division, differentiation of tissues, and maintenance of stem cell populations for tissue regeneration and repair [31].
Alterations in epigenetic modifications in the breast cancer genome probably occur relatively early in the oncogenesis process, possibly even before the process is recognized as such. These so called “epimutations” may therefore be initiators of carcinogenesis. Because of their early occurrence and broad-based, stable properties, epigenetic modifications have the potential to serve as useful biomarkers for tumorigenesis. These might allow earlier detection than other technologies. The consistent presence of epigenetic changes in malignant cells relative to normal also suggests that systems that control epigenetic processes may be suitable therapeutic targets in breast cancer. Indeed, histone deacetylases are being tested in triple negative breast cancer. Initial results suggest that HDACs may be able to reactivate expression of estrogen and progesterone receptors in TNBC, which might open new avenues to treatment of the disease [32].
9.3.7 Molecular Classification of Breast Cancer
All of the foregoing molecular knowledge on IBC is being brought together to form new classifications for the disease that better describe the biology and assist in therapeutic decisions. The development of DNA microarrays to measure RNA expression from large numbers of genes simultaneously (gene expression profiling) allowed hypotheses regarding the effects of genetic alterations on phenotype to be tested. Conceptually, the fundamental phenotype of cancer cells is reflected in their collective range of gene activity. Gene expression profiling for cancer measures the expression of hundreds or thousands of genes and can tie phenotype more closely to the essential defects in the malignant cell than can visual observation or a handful of biomarkers.
Perou, Sorlie, and colleagues, in their landmark publications [33–35], considered these patterns of gene expression as “portraits” of IBC cells. Computational methods were used to group gene expression profiles from large numbers of breast tumors to identify common patterns of gene expression that correlate with phenotype. Their goal was to develop an improved taxonomy for breast cancer that might more accurately reflect the clinical behavior and response to therapy of the tumors.
Use of hierarchical clustering methods resulted in identification of five related patterns of gene expression: the so-called “intrinsic subtypes” (Table 9.3). Two of these, Luminal A and Luminal B, resembled gene expression patterns identified in luminal-facing ductal epithelial cells which are usually ER+ and PR+. The HER2-enriched group typically expresses elevated levels of HER2 and is ER-, while the Basal group is characterized by lack of expression of all three of these genes. Although the Basal subgroup is often triple negative, upon closer examination not all Basal tumors are triple negative and not all triple negative tumors have a Basal gene expression pattern. The fifth subgroup initially identified by Sorlie and colleagues was termed the Normal-like subtype. The exact definition of these subtypes continues to be debated, however, the basic classification scheme originally described has been independently validated several times. These intrinsic subtypes have been shown to reflect the clinical behavior of the tumors and, therefore, have prognostic value. Because this classification is repeatable and clinically relevant, it has been incorporated into other breast cancer studies along with more traditional biomarkers [36].
Table 9.3
Properties of breast cancer intrinsic subtypes
Intrinsic subtype | Characteristics |
|---|---|
Luminal A | Gene expression pattern resembles ductal epithelial cells. Expression of cytokeratins 8 and 18. Typically ER+ and PR+ |
Luminal B | Shares most gene expression features with Lum A, but lower ER expression and expression of genes characteristic of HER2-Enriched subtype, although no overexpression of HER2 |
HER2 enriched | Overexpression of HER2 (ERB-B2) and genes nearby on chromosome 17 |
Basal | Gene expression pattern resembles myoepithelial (basal) cells of the duct. Expression of cytokeratins 5/6 and 17. Typically ER- and PR- |
Normal-like | Expression of genes known to be expressed in adipose and other non-epithelial cell types, e.g. fatty-acid binding protein 4 and PPAR gamma |
Several other gene-based classification methods have been develop and are in clinical use, including the widely used Oncotype DX Breast Cancer Assay [37]. The philosophy behind the development of these tests is the same as that for the intrinsic subtypes: develop assessments that more accurately measure the underlying biology of the tumor to more accurately predict clinical course and response to treatment. The strategy used to develop several of these tests identified genes with expression that is altered in cancer as compared to normal breast tissue. In principle, these gene expression changes may be tied more closely to the malignant phenotype than intrinsic subtypes. In the case of Oncotype DX and several others, RNA abundance measurements from a defined set of genes in tumor tissue are used to calculate a “risk score” that correlates with clinical behavior, usually the probability of distant recurrence.
Other gene-based tests have been developed and are now commercially available that measure protein abundance or epigenetic changes rather than changes in RNA concentration. Mammostrat® measures the abundance of five proteins (p53, HTF9C, CEACAM5, NDRG1, and SLC7A5) on tissue microarrays; an algorithm incorporating the expression levels is used to stratify patients according to risk level [38]. Although none is available for breast cancer currently, a test that measures promoter methylation in DNA from fecal samples is available to detect colon adenomas and tumors [39].
9.3.8 Tumor Microenvironment
9.3.8.1 Angiogenesis
Angiogenesis is a tightly controlled normal physiologic process that tumors take advantage of for their growth [40]. The normal angiogenic process does not occur in tumors, but rather the growth and development of disorganized, leaky vessels that serve as the vasculature of the tumor. Without this vasculature, despite its deficiencies, tumors are limited in their growth potential.
Because of the necessity for a blood supply for tumors to grow appreciably, angiogenesis is a natural target [41]. Early research into angiogenesis identified a number of proteins dedicated to the process. Notable among these are vascular endothelial growth factor (VEGF) and its receptor, which together regulate new vessel formation. A humanized monoclonal antibody directed at VEGF-A, bevacizumab, has been extensively tested as a therapeutic in IBC. Bevacizumab was approved for adjuvant therapy of metastatic breast cancer based on results of large registration trials. However, post-market testing of bevacizumab did not support efficacy in the general population of patients with metastatic breast cancer. As a result, the US FDA withdrew its approval for this indication. It is now accepted that pathways regulating angiogenesis are redundant, complicating the use of single anti-angiogenic agents. Multiple alternative anti-angiogenic agents targeting other pathway components are in development [42].
9.3.8.2 Inflammation and Tumor Immunology
Evidence of inflammation and the presence of immune cells in tumors was initially considered a sign of effective host response on the tumor. It is now recognized that immune cells can also promote cancer initiation, progression, and metastasis. For example, tumor associated macrophages (TAMs) are recruited by tumors through secretion of cytokines including CCL2 and colony stimulating factor 1. TAMs have been found to have significant pro-tumor effects by supporting angiogenesis, suppressing immunity, and enhancing migration [43]. Targeting of these macrophages can have significant antitumor effects. Other inflammatory pathways have been shown to have pro- or anti-tumor effects that can be modulated. These include tumor growth factor beta (TGF beta), NF kappa B, IL6-/JAK/STAT, tumor necrosis factor alpha (TNF alpha), and COX2 signaling pathways. Manipulation of these pathways will yield additional therapeutic strategies.
Similarly, it is been discovered that T and B lymphocytes can also be pro- or anti-tumorigenic. As the details of the immune cell activation process have been worked out, it has become apparent that it is being triggered by tumors. However, it has also been discovered that specific aspects of the interaction of the tumor with the T cells appear to cause co-repression of the immune response, which may contribute to malignant cells evading immune attack [44, 45].
New strategies are being developed to reverse this co-repression. A key element of the activation of the T cell response to antigen is accomplished is by interaction of the T cell receptor (TCR) with antigen peptide in the context of major histocompatibility complex (MHC) bound antigen on antigen presenting cells (APC). For this signal to be effective, co-stimulation must accompany the interaction. Proteins of the CD28/B7 family interact to provide this co-stimulation. To provide for control of the immune response, this stimulatory signal must be balanced with an inhibitory signal. Other proteins of the CD27/B7 family provide this regulatory co-inhibitory signaling, including CTLA-4 and PD1/PD-L1. In breast and other cancers, the balance between co-stimulation and co-inhibition appears to be skewed toward co-inhibition by dysregulated expression of several of the B7 and CD28 family members on tumor cells. Recent clinical studies using antibodies to block the co-repression appear to have a significant anti-tumor effect.
Several other inflammation-related pathways may be ineffective or aberrantly active in breast cancer, including NF kappa B signaling, TNF alpha signaling, and IL-6/JAK/STAT3 signaling. Strategies for intervening in these pathways are still under development [42].
9.3.8.3 Communication with the Bone Microenvironment
Bone is a primary metastatic site for many epithelial tumors, including breast cancer. Parathyroid hormone-related peptide (PTHrP) from tumor cells stimulates differentiation of precursor cells into osteoclasts via RANKL expression in osteoblasts. This process is inhibited by osteoprotegerin. Interference with this process is a strategy for targeted therapy of bone metastases. In addition, bone stroma secretes TGF beta, which can stimulate tumor cell growth [46].
To date, two drugs that target osteoclasts to address morbidity from bone metastases have been approved. The bisphsophonate, zoledronic acid, significantly reduces skeletal related events (SRE) in patients with boney metastases. Denosumab is a humanized monoclonal antibody that targets RANKL and is approved to prevent SREs in patients with solid tumors. The FDA has also approved denosumab to increase bone mass in breast cancer patients receiving adjuvant aromatase therapy.
9.3.9 Molecular Medicine
The objective of most therapeutic research in cancer medicine is to more specifically target the malignant disease process, sparing normal cells and tissues. The pursuit of improved “therapeutic index” has driven cancer therapy research and development for more than 60 years. Development of new therapeutic methods was guided by available evidence and hypotheses regarding disease mechanisms that were based on distinctive features of the disease that were observable at the time. For example, recognition that a primary observable feature of leukemia was excessive cell division led to use of alkylating agents, which selectively attack dividing cells, as systemic therapies for cancer. While the hypothesis was sound based on what was known (the primary characteristic of leukemia is excessive division), high levels of systemic toxicity were observed because of bystander effects on normal dividing cells. Thus, the therapeutic index was narrow.
Since that time, progress in cancer therapy development, particularly for breast cancer, has gratifyingly yielded therapies that are much more specific for the malignant cell. The burgeoning understanding of the similarities and differences between normal breast epithelial cells and malignant breast epithelial cells and their environment has helped form the basis for developing the concepts that comprise molecular medicine (the same principles apply to other normal and corresponding malignant cell types). As one of the most active areas of research in biomedicine, breast cancer research has benefitted from several molecular medicine developments, both diagnostic and therapeutic, that have made the transition from the laboratory to the clinic.
9.4 Diagnosis
New primary breast tumors often present as palpable masses, persistent areas of pain or tenderness, nipple discharge, or as suspicious radiographic findings. Within the last several years, the recommendations regarding breast cancer screening have shifted. It had previously been recommended that clinicians teach women how to perform regular breast self-examinations; however, a 2008 Cochrane metaanalysis based on two randomized controlled trials including nearly 390,000 women in China and Russia led to a change in practice. In this review, women with regular breast self-exams had no improvement in breast related survival but did have more invasive procedures for benign lesions [47]. In part based on these data, the U.S. Preventative Services Task Force recommended that clinicians no longer teach patients to perform breast self-exams [48]. In recent years there has been a large increase in the number of breast abnormalities detected radiographically due to wide-spread screening mammography. For tumors identified initially as palpable abnormalities the next steps toward a definitive diagnosis are imaging studies, followed by biopsy and histopathology studies, while those detected using mammograms generally proceed directly to biopsy and tissue studies.
9.4.1 Imaging
9.4.1.1 Mammography
Mammography has a long-standing record as a diagnostic imaging technique to investigate suspicious lesions. Calcifications, distortion of tissue architecture, and other signs on mammography suggest that biopsy of the lesion is necessary.
Because of its success as a diagnostic tool, low cost, and non-invasive nature, mammography is now used as a screening tool for breast cancer. As with other screening technologies the balance between imperfect sensitivity and imperfect specificity means that in order to identify the desired number of patients with disease early in their course when intervention is most effective, some number of individuals without meaningful disease will be called back. Normally, for cancer the balance is tilted toward accepting some level of false positives. Considering other risk evaluation criteria, such as age, in combination with the radiographic results assists in maximizing the utility of mammography. Additional sensitivity and specificity for accurate diagnosis in mammography can be achieved by trained and experienced mammographers using quality systems with appropriate support. To aid clinicians in interpreting the results of mammography reading, the Breast Imaging-Reporting Data System (BI-RADS) classification system was developed and is standard in mammography reporting. Lesions are graded from 0 to 6 to help guide the management of radiographic breast lesions (Table 9.4).
Table 9.4
BI-RADS classification of mammographic breast lesions
BI-RADS classification | Assessment |
|---|---|
0 | Incomplete study, consider additional or repeat imaging or obtain previous imaging for comparison |
1 | Negative study; continue routine screening mammography |
2 | Benign findings; continue routine screening mammography |
3 | Probably benign findings; repeat mammography in 6 months |
4 | Suspicious lesion—biopsy recommended |
5 | Highly suspicious for malignancy—biopsy recommended |
6 | Lesion is a known biopsy-proven malignancy |
Several long-term randomized controlled trials of population-based screening for breast cancer by mammography have demonstrated a reduction of between 28 % and 45 % in disease-specific mortality [49]. A systematic review by the U.S. Preventative Services Taskforce confirmed that mammography reduces breast cancer mortality in women between 39 and 69 years old [50]. The benefit of screening was maximal for women ages 50–74. Although the mortality benefit of mammography is not disputed, there are controversies with respect to the interval used for screening, as well as the benefit for younger women. Current guidelines in the U.S. suggest biennial mammography for women 40 or over.
9.4.1.2 Other Imaging Modalities
Once a malignant breast tumor is suspected or proven by biopsy, additional imaging studies may be warranted to assess the extent of disease, as well as evaluate additional characteristics of the tumors, such as size, location, and proximity to other structures. Additional imaging studies also have the advantage of being less invasive, less painful, and less expensive than other assessments, including biopsy, which may subsequently be needed. Clinically available technologies include ultrasonography (US), magnetic resonance imaging (MRI), positron emission tomography/computerized tomography (PET/CT), and scintigraphy [51].
US is simple to perform and readily available. This technique can help to quickly distinguish cystic disease from tumors and also to gather additional information about the lesion, but at the cost of a fairly low sensitivity. MRI is highly sensitive and allows evaluation of multiple sites to resolve questions that cannot be answered using mammography or US. However, MRI has limited availability and its expense is an issue in the current cost-constrained medical environment. PET/CT can be a sensitive method to detect disease not easily visualized by other methods. This technology can also provide metabolic information by quantitating the uptake of labeled glucose tracer. However, as with MRI, PET/CT has limited availability, is cumbersome, and exposes patients to radiation. Scintigraphy using 99Tc methylene diphosphonate (MDP) can sensitively detect metastases to the skeleton, a common occurrence in breast cancer. The specificity of bone scans for malignant disease is lower, however, since osteoblastic activity is detected, which may not be related to breast cancer. This technology is widely available and is safe.
9.4.2 Biopsy
The gold standard for diagnosis of breast cancer is examination of tissue specimens. A lesion that has been identified as potentially malignant by clinical and radiographic assessments must be biopsied to establish a definitive diagnosis of breast cancer [52].
Three modes of biopsy are typically employed (Table 9.5). Fine needle aspiration (FNA) is quick, inexpensive, and relatively painless. The tissue sample can be used to identify morphologically abnormal cells, however, invasiveness of the lesion and expression of multiple biomarkers cannot be assessed given the small amount of tissue. In addition, a pathologist trained in interpretation of FNA samples must perform the evaluation.
Table 9.5
Breast lesion biopsy techniques
Technique | Advantages | Disadvantages |
|---|---|---|
Fine needle aspiration (FNA) | Minimally invasive, relatively painless, in-office procedure, inexpensive | No histological evaluation possible, requires specialized expertise, biomarker analysis not usually feasible |
Core needle biopsy | Minimally invasive, relatively painless, in-office procedure, inexpensive, no specialized pathology expertise needed, biomarker analysis possible | Possible false-negatives, incomplete lesion evaluation |
Excisional biopsy | False negative results rare, complete lesion evaluation, may serve as definitive lumpectomy | More expensive, painful, surgery may be unnecessary if lesion is benign |
May require further breast excision if malignant disease is found |
Core biopsy using a core needle provides a larger sample suitable for histologic evaluation, enabling any pathologist to perform the evaluation. Biomarker analysis can routinely be performed on core biopsies. As with FNA, sampling errors can cause false negative results. Concordance between biopsy, clinical evaluation, and imaging is important; in the absence of concordance additional tissue should be sampled.
Excisional biopsy is the most invasive sampling procedure, but may also serve as the definitive lumpectomy in some cases. A small margin of normal tissue should be obtained, orientation sutures should be placed, and surfaces should be inked to allow follow-up surgery to be performed with minimal additional trauma.
Diagnosis by core needle biopsy is the preferred method for evaluating almost all breast masses. This procedure usually enables discussion of all therapeutic options prior to embarking on potentially more invasive options.
9.5 Pathology
9.5.1 Introduction
The breast is a highly developed, highly endocrine sensitive organ in females versus a vestigial, endocrine insensitive organ in males. The organ itself is composed of epidermal, dermal, breast stromal, and breast glandular tissues. The glandular tissue comprises approximately 10–15 % of the tissue by volume and is the site of origin of virtually all breast cancers. The glandular tissue is divided into 15–20 lobes, with the space between lobes filled with connective tissue and adipose tissue. The vasculature of the breast is derived from the internal mammary artery and the lateral thoracic artery. The lymphatic drainage connects to a superficial and a deep plexus, with more than 95 % of the drainage directed toward the axillary lymph nodes [52].
The ductal system of the breast consists of the nipple, lactiferous ducts, segmental ducts, and terminal duct lobular units (TDLU, Fig. 9.2). The components of the TDLU are the terminal duct and the lobule. The TDLU is the location where most carcinomas arise. The lobule consists of ductules and acini. Underlying these components is a basement membrane on which a layer of myoepithelial cells rests. Further toward the lumen is a layer of columnar epithelial cells. The organization of these ductal tissue bears on the pathological assessment, with intact basement membrane demarcating in situ disease with its distinct prognosis.
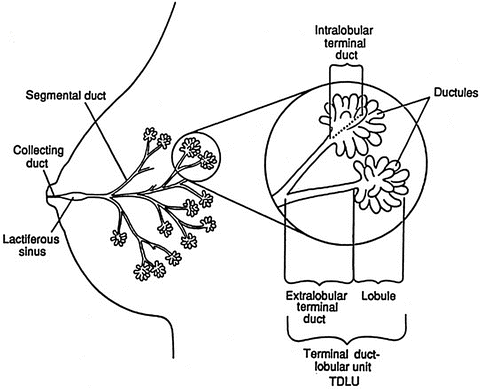
Fig. 9.2
Normal breast anatomy. The normal ductal anatomy is shown. The inset is a magnified view of the structure of the terminal ductal lobular unit (TDLU) (Reproduced from [53] with permission)
9.5.2 Pathologic Assessment
When breast cancer is suspected, the objective for the pathologist is to make the distinction between invasive breast cancer, in situ disease, and other non-malignant proliferative lesions of the breast. There are a variety of non-malignant conditions that may produce masses resembling tumors in some way. In one study of a large series of women with breast complaints, 40 % had fibrocystic disease, 10 % had biopsy-proven malignant tumors, and 7 % had benign tumors.
Standard Pathologic Management
Diagnosis of invasive breast cancer
Exclusion of benign proliferative disease and in situ disease
Characterization of nuclear morphology, tumor architecture, mitotic activity
Tumor size and extent (locally advanced or metastatic)
Surgical margins (early stage or excisional biopsy)
Determination of biomarker status (ER, PR, HER2)
Staging
Criteria used to make the histopathologic diagnosis include nuclear morphology, mitotic activity, and tissue architecture. From this information a classification as malignant or not and, if applicable, malignant classification can be made. The most common classifications are invasive ductal carcinoma (IDC), ductal carcinoma in situ (DCIS), and invasive lobular carcinoma (ILC). Additional information required for complete pathological assessment includes, surgical margins, nodal status, and biomarker expression. Each of these parameters contributes to staging, treatment planning, and ultimately prognosis.
The major histologic criteria that distinguish IDC, ILC, and DCIS are nuclear morphology, stromal and ductal element architecture, and basement membrane integrity. The “ductal” and “lobular” designations in histopathologic diagnoses do not indicate location of origin, but rather morphologic type [54].
IDC includes 70–80 % of breast carcinomas and includes those that cannot be classified as any other subtype. Rather than being a distinct morphological type of cancer, IDC is equivalent to the Not Otherwise Specified (NOS) or of No Special Type (NST) designation used in the classification of other cancers. Histologically, most have abundant fibrous stoma that gives the tumor a firm consistency, also called a scirrhous carcinoma. Tumor cells are seen to be invading the stroma in cords or nests. The nuclear morphology ranges from moderately hyperchromatic to large, irregularly shaped, and very hyperchromatic.
DCIS is differentiated from invasive cancer by the presence of an intact basement membrane and a circumferential and intact myoepithelial cell layer. With the advent of mammographic screening for cancer, DCIS as a diagnosis has increased tremendously and now comprises about 20–25 % of newly diagnosed breast cancers. Five types of DCIS are recognized based on histologic architecture: comedo, solid, cribriform, papillary, and micropapillary. Usually some mixture of these patterns is observed.
Invasive lobular carcinoma contributes 5–10 % of breast carcinomas. Histologically, the tumors have a diffusely invasive pattern which may make the tumors more difficult to detect by palpation or mammography. The cells are small with less nuclear pleomorphism than IDC. Cells are usually diploid and HR positive. The prognosis for ILC is better than for IDC. The remaining proportion of cases are composed of a mix of several different types of less common tumors.
Various grading systems have been developed over time in recognition of the fact that diminished differentiation of tumor cells, reflected in histologic features of the tumor, correlate with reduced survival. Currently, the most commonly used system is the Nottingham System, modified from the Bloom Richardson grading system [55]. The grade of cancer is representative of the aggressive potential; low grade cancers tend to be less aggressive than high grade cancers. The Nottingham Histologic Score system uses three factors to arrive at grade:
The amount of gland formation
Nuclear features
Mitotic activity
The features receive a score from 1 to 3, which are then summed to yield a final grade score ranging from 3 to 9. Grade 1 tumors have a score of 3–5, grade 2 tumors have a grade of 6–7, and grade 3 tumors have a score of 8–9. Grade 3 tumors are the most aggressive and carry the worst prognosis (Fig. 9.3).
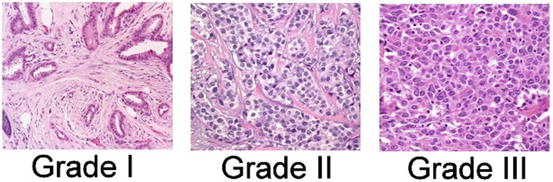
Fig. 9.3




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
