Childhood interstitial lung disease (ChILD) is an umbrella term encompassing a diverse group of diffuse lung diseases affecting infants and children. Although the timely and accurate diagnosis of ChILD is often challenging, it is optimally achieved through the multidisciplinary integration of imaging findings with clinical data, genetics, and potentially lung biopsy. This article reviews the definition and classification of ChILD; the role of imaging, pathology, and genetics in ChILD diagnosis; treatment options; and future goals. In addition, a practical approach to ChILD imaging based on the latest available research and the characteristic imaging appearance of ChILD entities are presented.
Key points
- •
Childhood interstitial lung disease (ChILD) is an umbrella term referring to a diverse group of diffuse lung diseases occurring in childhood.
- •
ChILD differs in appearance from adult ILD with common, important confounding features that must be recognized, which include respiratory motion mimicking ground glass opacification and reversible “bronchiectasis” in the context of acute or recent viral infection.
- •
Computed tomography (CT) remains the current gold standard imaging modality for the confirmation and characterization of suspected ChILD after chest radiography because of its high sensitivity and increased likelihood of providing a specific diagnosis.
- •
Optimal CT technique is crucial to ChILD imaging with diagnostic-quality CT images often possible in free-breathing nonsedated infants.
- •
A clear understanding of the characteristic imaging findings of certain ChILD entities is imperative for timely diagnosis and optimal patient management.
Introduction
Childhood interstitial lung disease (ChILD) is an umbrella term encompassing a diverse group of diffuse lung diseases affecting infants and children. Although the timely and accurate diagnosis of ChILD is often challenging, it is optimally achieved through the multidisciplinary integration of imaging findings with clinical data, genetics, and possibly lung biopsy. This article reviews the definition and classification of ChILD; the role of imaging, pathology, and genetics in ChILD diagnosis; treatment options; and future goals. In addition, a practical approach to ChILD imaging based on the latest available research and the characteristic imaging appearance of ChILD entities are presented.
Childhood interstitial lung disease definition and classification
The term interstitial lung disease (ILD) refers to a large, heterogeneous group of diffuse parenchymal lung diseases. In adult practice, these conditions are generally well characterized. Adult ILDs are broadly divided into 3 groups: (1) the major idiopathic interstitial pneumonias such as idiopathic pulmonary fibrosis (IPF), nonspecific interstitial pneumonia (NSIP), organizing pneumonia, etc; (2) the rare idiopathic interstitial pneumonias such as lymphocytic interstitial pneumonia (LIP) and pleuroparenchymal fibroelastosis (PPFE); and (3) the unclassifiable idiopathic interstitial pneumonias.
In adult ILD, the diagnostic emphasis is on distinguishing fibrotic from inflammatory entities, in particular distinguishing the specific diagnosis of IPF—defined by the histologic pattern known as usual interstitial pneumonia (UIP)—from other ILD diagnoses. The underlying need for this distinction is based on well-established evidence of differences in prognosis and, more recently, differences in treatment of each condition.
A comparative study published in 2000 is an important example of the evidence required to advance adult ILD care to its current state. Nicholson and colleagues reviewed the histopathology specimens from 78 patients diagnosed with cryptogenic fibrosing alveolitis (CFA) in the late 1970s to the 1980s. CFA is a now an outdated term more recently subdivided into separate specific entities, and as such Nicholson and colleagues reclassified these patients into UIP, NSIP, and desquamative interstitial pneumonia/respiratory bronchiolitis-associated interstitial lung disease (DIP/RBILD, the smoking-related ILDs) diagnoses. More than 85% of the patients they examined had clinical follow-up to death or to 10 years postbiopsy, with significantly higher mortality demonstrated in UIP than NSIP ( P <.01), no deaths in the DIP/RBILD group, and more frequent treatment response in DIP/RBILD than in either NSIP or UIP ( P <.01 and <.001, respectively). This distinction in diagnosis and the accompanying knowledge of prognosis and expected treatment response is of critical value to those involved in adult ILD care.
Unfortunately, the knowledge and evidence base available to those working with ILD in the pediatric population is a very different story. Childhood ILD (neatly abbreviated as ChILD) is an umbrella term encompassing more than 200 rare diffuse lung diseases that affect children. Although variably reported, the incidence of ChILD is less than 1 per 100,000 population, 60 to 80 times less frequent than adult ILD. , Although a few specific ILD diagnoses are common to both adults and children, the disease severity and clinical course are often different and there are many ILD diagnoses unique to the pediatric population. As such, the classification of ChILD differs from adult ILD classification systems. In 2007, Deutsch and colleagues devised a classification system distinguishing between ChILD entities based on patient age at presentation, dividing conditions into those that generally present in infancy and those that are not specific to infancy ( Box 1 ).
- 1.
Disorders more prevalent in infancy
- a.
Diffuse developmental disorders
- b.
Lung growth abnormalities
- c.
Specific conditions of undefined etiology
- d.
Surfactant dysfunction mutations and related disorders
- a.
- 2.
Disorders not specific to infancy
- a.
Disorders of the normal host
- b.
Disorders related to systemic disease processes
- c.
Disorders of the immunocompromised host
- d.
Disorders masquerading as interstitial lung disease
- a.
- 3.
Unclassified
This classification system was revisited and recommended for routine use by the 2013 American Thoracic Society (ATS) clinical practice guidelines. Griese and colleagues tested the classification system by assigning diagnoses to 100 randomly selected patients from the “Kids Lung Register,” the registry of the European ChILD-EU initiative. Cases were initially classified based on a multidisciplinary team of subspecialty pediatric pulmonologists, radiologists, and pathologists. Reclassification of these same patients several years later by 2 pediatric pulmonologists demonstrated more than 80% agreement with initial diagnoses. Just as in adult ILD practice, this multidisciplinary approach to ChILD diagnosis is critical to accurate and reproducible classification of individual ChILD diagnoses, requiring integration of all available information from clinical history, laboratory tests, imaging, bronchoalveolar lavage (BAL), and lung biopsy. This fact is highlighted by a study by Jacobs and colleagues in which experienced subspecialty expert ChILD radiologists and pulmonologists categorized 84 ChILD cases into broad groups and then more specific etiologies, based solely on imaging appearance with no further clinical information. Compared with the 80% agreement in the multidisciplinary study of Griese and colleagues, this CT-alone approach yielded, at best, fair interobserver agreement in diagnosis, with Fleiss kappa values ranging from 0.16 to 0.44. Consequently, a multidisciplinary approach to ChILD classification is critical for the accurate diagnosis and understanding of disease processes.
The role of imaging, genetics, and pathology in childhood interstitial lung disease diagnosis
The 2013 ATS guidelines present 3 diagnostic algorithms, 2 for infants presenting with severe respiratory disease in the neonatal period (altered depending on the presence or absence of a family history of ILD) and 1 for children presenting after 1 month of age. All three algorithms for ChILD diagnosis feature imaging early in the diagnostic pathway, as soon as more common non-ChILD diagnoses (infection, cardiovascular disease, immunodeficiency, cystic fibrosis, and aspiration) have been excluded.
Genetic testing is a more recent addition to the multidisciplinary ChILD diagnostic process. Genetic testing is of increasing importance, particularly when ChILD presents in infancy and even more so when there is a strong family history. The aim of both imaging and genetic studies is to arrive at a specific diagnosis without invasive testing, with each algorithm ending with surgical lung biopsy if no specific diagnosis can otherwise be reached.
Even if imaging does not lead to a specific ChILD diagnosis, its role in multidisciplinary practice remains a key factor. Many diffuse lung diseases demonstrate nonhomogeneous lung involvement with geographic regions of sparing or differing disease patterns. Consequently, prebiopsy imaging is vital to guide the surgeon to an area of involved lung and in the subsequent interpretation of biopsy results.
A 2004 study by a European Respiratory Society task force identified ChILD cases across Europe, examining among other things, the success of the diagnostic pathway. Of 185 patients, a specific diagnosis was reached in 177 (96%), with diagnosis reached without surgical biopsy in 78 (45%). The success of noninvasive diagnosis in any cohort clearly depends on the prevalence of individual disease entities contained therein. In addition, one major, albeit understandable, limitation of ChILD research to date is the paucity of studies into the diagnosis and management of individual ChILD entities, with many studies investigating the prognosis and treatment responses of ChILD as a single entity.
Treatment options and future goals
Because such a broad variety of entities fall under the umbrella term ChILD, it is not helpful to consider treatment of ChILD as a single diagnosis. However, the rarity of individual diagnoses and the relatively recent application and availability of genetic studies severely limits the ability to reach the level of evidence-based practice seen in adult ILD care. Outside of a few specific ChILD disorders with specific treatments (mostly those with rheumatologic associations), the mainstay of ChILD treatment includes reduction in inflammation with corticosteroids, hydroxychloroquine, and azithromycin; supportive care with oxygen, noninvasive and invasive ventilation, extracorporeal membrane oxygenation (ECMO), and transplantation; and preventative measures, such as management of chronic aspiration and/or reflux, routine immunizations, and the avoidance of exposure to air pollutants. ,
Future imaging goals include larger, multicenter, and international cohort studies of the imaging features of specific individual ChILD entities, complete with extensive clinical characterization (including detailed genetic analysis), to enable better understanding of the clinical significance of differing imaging phenotypes. Any cohort study across international borders is likely to include numerous different scanners and different scan protocols, and to attempt to standardize computed tomographic (CT) imaging for ChILD would likely result in a “race to the bottom” with suboptimal imaging quality on older scanners and far higher than necessary doses on newer scanners. Rather, a focus on site-specific tailoring of CT protocols, with input from medical physics experts and routine image quality and dose audit, is likely to result in appropriate diagnostic quality imaging being achieved. Furthermore, it can subsequently enable analysis and comparison across centers. ChILD-specific organizations, such as ChILD-EU and the US Children’s Interstitial and Diffuse Lung Disease Research Network (ChILDRN), are likely to play a key role in enabling sufficient cross-border collaboration for the formation of sufficiently large patient cohorts.
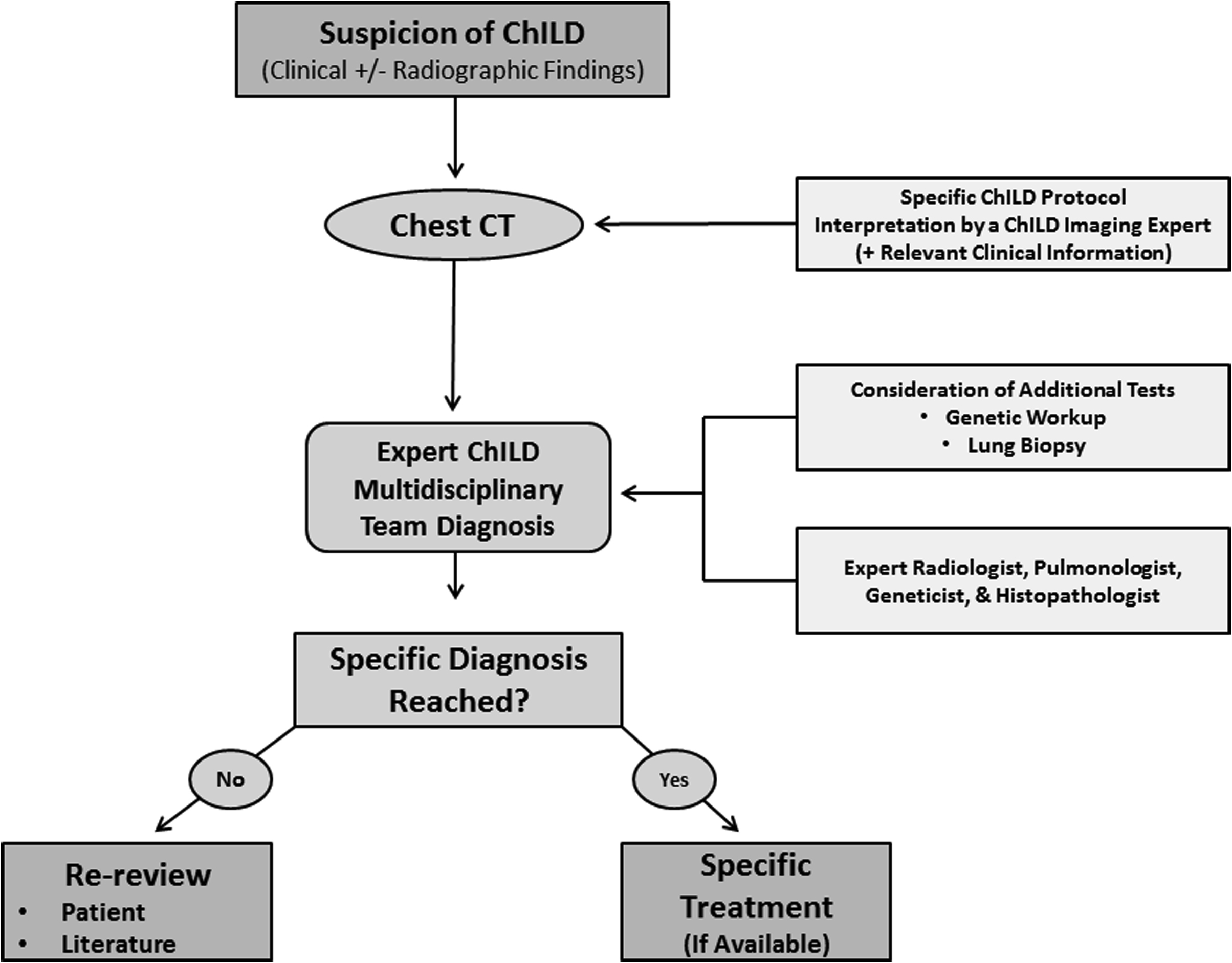
A review of imaging technique and of the imaging features of specific ChILD disorders follows. Based on the aforementioned facts, our suggested algorithm for the investigation of suspected ChILD is presented in Fig. 1 .
Practical imaging approach to pediatric interstitial lung disease
Various imaging modalities are currently available with radiography and CT as the 2 most important and widely available imaging modalities for evaluating ChILD. A brief summary of their current roles along with relevant advantages and disadvantages are discussed in the following section.
Radiography
The chest radiograph is the most commonly performed respiratory imaging investigation worldwide, providing a single image overview of the entire thorax. In both children and adults, the sensitivity of chest radiography to ILD is lower than that of CT, with normal radiographic appearances of ChILD at initial presentation variably reported in 10% to 42% of children with later proven ILD. , However, it is often the radiographic appearance that leads to further more sensitive and specific cross-sectional imaging evaluation of ChILD. , In clinical practice, it is rare to encounter a request for CT for suspected ChILD without a prior chest radiograph for reference. Although less detailed than CT, a diagnostic-quality chest radiograph can provide an easily available, low dose form of follow-up imaging without the higher ionizing radiation exposure that would be required for frequent follow-up via CT. Therefore, chest radiography should be reviewed before initial CT assessment and considered as the first-line follow-up imaging modality in the absence of substantial clinical decline of children with ILD.
There are several clinically relevant diagnoses that can be made on chest radiography in the setting of suspected ChILD. For example, when the diffuse granular appearance of typical surfactant deficiency of prematurity is seen in a term neonate, this may suggest underlying congenital surfactant dysfunction (eg, SPFB or adenosine triphosphate-binding cassette transporter A3 [ABCA3] mutations). In premature infants, chronic lung disease of prematurity, previously known as bronchopulmonary dysplasia (BPD), has an almost pathognomonic radiographic appearance characterized by alternating foci of hyperinflation and atelectasis, nearly as readily appreciable on chest radiography as on CT. Another radiographic appearance worth mentioning is that of atelectasis and coarse peribronchial thickening with a bilateral lower lobe and right upper lobe distribution, an appearance often encountered in the setting of chronic aspiration. Aspiration alone can present as respiratory distress mimicking ChILD, but perhaps more clinically importantly, feeding issues are reported in as many as 73% of patients with a ChILD diagnosis with 35% requiring long-term gastrostomy feeding and 14% reporting oral aversion suggesting the possibility of chronic aspiration. If undiagnosed and/or left untreated, chronic aspiration is a substantial additional risk to patients with ChILD.
Given the aforementioned facts, the roles of chest radiography in ChILD assessment include (1) initial suggestion of a ChILD diagnosis, (2) primary evaluation of disease distribution and appearance before CT imaging is available, (3) investigation of extrapulmonary features of specific ChILD entities, (4) detection of features of chronic aspiration (as a primary or exacerbating pathologic condition), and (5) as a follow-up evaluation imaging modality (ideally as part of a routine assessment akin to the annual evaluation in cystic fibrosis care). It is important to recognize that, given the reduced relative sensitivity and specificity, a normal chest radiograph should not be seen as excluding ChILD and CT should always be performed where there is ongoing clinical suspicion in the clinical setting of persistent or worsening symptoms.
Computed Tomography
As discussed previously, the superior anatomic detail provided by CT in combination with utilization of multiplanar reformats and 3D reconstructions leads to a far higher sensitivity for the diagnosis of ChILD compared with chest radiography. Furthermore, if lung biopsy is required, CT can guide the surgeon to an appropriate biopsy site, limiting the possibility of a nondiagnostic or nonrepresentative biopsy sample. ,
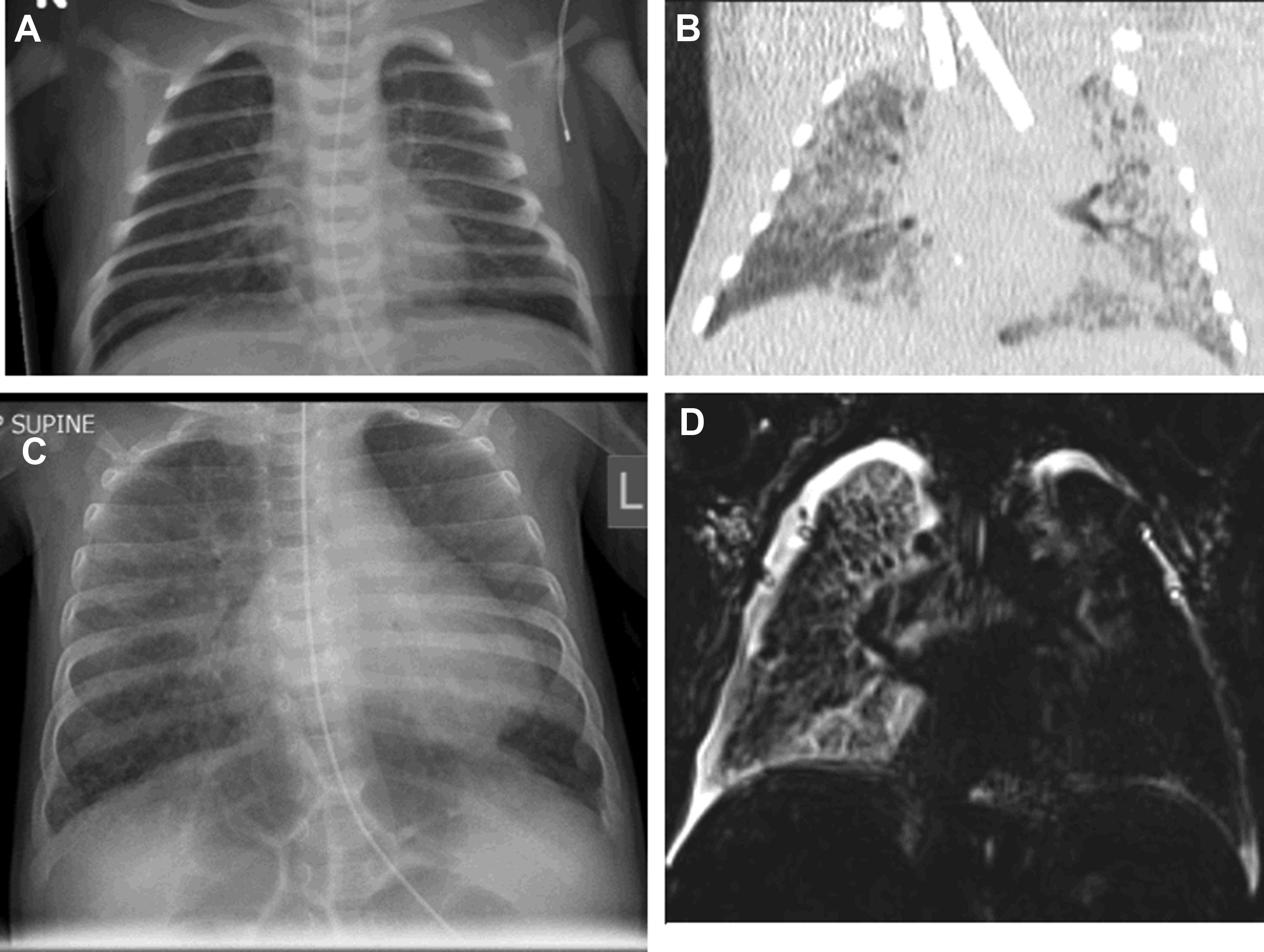
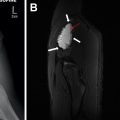
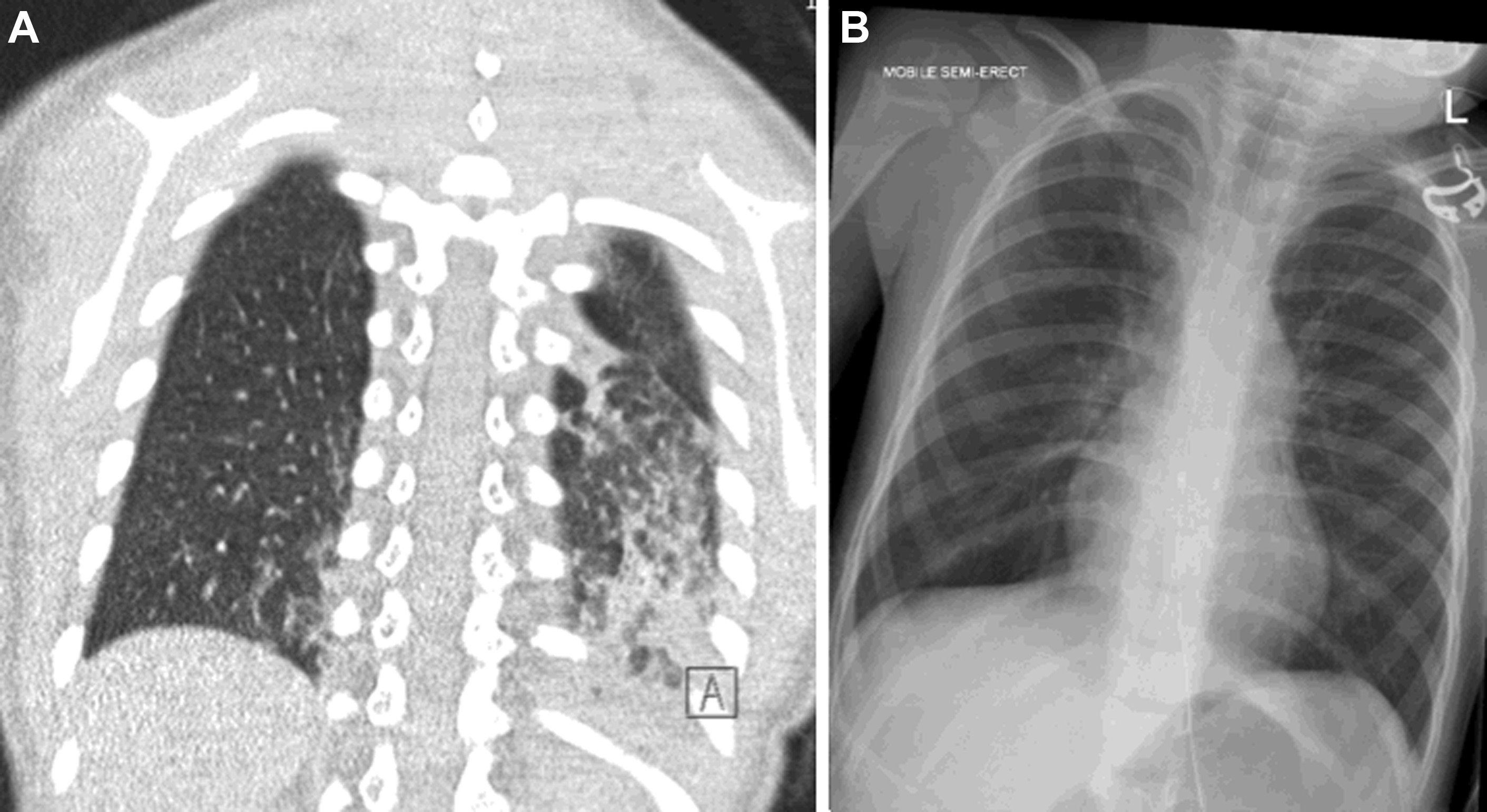
While the ATS guidelines emphasize the important role of CT as the gold-standard imaging modality in ChILD diagnosis, there have been some advances, particularly in CT technique. The ATS guidelines recommend the use of “controlled ventilation high-resolution CT,” with deep breaths delivered to a sedated child via a mask; this results in a short period of apnea during which imaging can be acquired with minimal respiratory motion. Modern CT scanners are now far faster than their historic equivalents with top-end CT scanners capable of imaging the entire thorax of an adult in a fraction of a second. In infants and small children, this means that it is now possible to acquire diagnostic-quality lung imaging during free breathing, without the need for sedation. It is well known that sedation and anesthesia result in the rapid accumulation of atelectasis in the dependent portion of the lungs, obscuring underlying pathologic conditions and mimicking the appearance of chronic aspiration ( Fig. 2 ). Awake, free-breathing imaging can eliminate this risk and ensures that the resultant CT images demonstrate the lungs in their normal physiologic state.
If anesthesia is required, for example, in noncompliant patients, typically age 2-5 years old or those with developmental delay, or when utilizing a CT scanner that is not capable of sufficiently rapid imaging), the use of lung recruitment maneuvers before CT imaging is essential in reducing dependent atelectasis and maximizing the quality of the CT. At the Royal Brompton Hospital, if anaesthesia is required, a standard CT protocol for ChILD involves at least 4 slow lung inflations followed by a held inspiration at around 25 cm H 2 O at the time of inspiratory CT imaging.
The ATS guidelines also mention the use of additional prone and decubitus positioning. , , Decubitus imaging can be useful in producing expiratory imaging in noncompliant pediatric patients (the more dependent lung is relatively expiratory compared with the nondependant lung), and prone imaging is occasionally used to reduce dependent atelectasis at the lung bases from mimicking subtle subpleural ground glass opacification. The inclusion of expiratory imaging in ChILD CT protocols is a current topic of debate. In the context of constrictive/obliterative bronchiolitis (COB), for example, as a complication of prior adenovirus infection or as a manifestation of graft-versus-host disease (GVHD), expiratory CT imaging is useful in demonstrating subtle mosaic attenuation. However, if this appearance is demonstrated on the inspiratory CT images, additional expiratory CT imaging adds little more than extra radiation exposure and should be reserved for research settings where low attenuation is being quantified at a strictly controlled point in the respiratory cycle, ideally with the use of spirometer guidance. In general, if COB is not the expected diagnosis, the routine inclusion of expiratory imaging in ChILD protocols may not be necessary. Prone positioning is almost never required in pediatric ILD imaging.
Along with improvements in the speed of CT acquisitions, there have been substantial reductions in the radiation dose required for diagnostic CT imaging. Modern CT scanners use advanced forms of 3D tube current modulation, kilovolt selection, and reconstruction algorithms that produce diagnostic-quality CT images from doses that would result in nondiagnostic, heavily noise-affected CT images using conventional CT image reconstruction methods. This improvement has led to the advent of low- and ultralow-dose CT protocols for lung imaging. Although these low dose CT techniques are useful for clinical settings in which low-resolution imaging is appropriate (eg, cystic fibrosis, infection, lung cancer screening in adults), it is the high-resolution capabilities of CT that make it central to the diagnosis of ChILD. As such, it is appropriate to have a slightly higher radiation dose, higher diagnostic yield, and higher confidence protocol specifically for use in the initial diagnosis of ChILD (ie, a single diagnostic-quality CT is preferred to an initial low-dose nondiagnostic CT that must be repeated because of too low radiation dose or overinterpretation of apparent diffuse ground glass opacification due to increased noise ( Fig. 3 ). The Royal Brompton Hospital’s pediatric pulmonary CT protocols are included in Table 1 . CT scan parameters should be matched to individual CT scanners with the input of a local medical physics expert. The inclusion of these protocols in this article is merely intended to demonstrate a tailored approach to pulmonary CT protocols.

| Protocol | Parameters |
|---|---|
| Low dose | Free breathing 80 kV CareDose On: QrefmAs 80 Pitch 3 |
| 0–10 kg ILD | Free breathing 80 kV CareDose On: QrefmAs 150 Pitch 3 |
| 10–50 kg ILD | Breath hold if compliant, otherwise free breathing 100 kV CareDose On: QrefmAs 130 Pitch 3 |
| Adult ILD | Breath hold at full inspiration 120 kV CareDose On: QrefmAs 110 Pitch 0.6 |
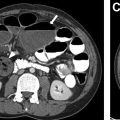
The goals of CT studies in children with suspected ChILD include confirmation of the presence of an ILD, demonstration of imaging findings of specific ChILD diagnoses, and guidance of potential future surgical biopsy. Specific diagnoses and their imaging appearances of ChILD are further discussed in the following sections; however, the imaging features of fibrotic disease bear mention here. In adult ILD, imaging features such as traction bronchiectasis, architectural distortion, volume loss (generally appreciated by displacement of the interlobar fissures), and the presence of honeycombing are generally accepted as being in keeping with the presence of a fibrotic ILD. Several key differences between adult ILD and ChILD disease processes and imaging features are important to keep in mind. The presence of acute infection at the time of imaging may result in the appearance of bronchial dilatation, thickening, ground glass opacification, and mosaic attenuation, closely mimicking the appearance of COB ( Fig. 4 ). Therefore, it is crucial to avoid diagnostic imaging for the evaluation for ChILD during times of acute infection if the imaging is specifically intended to characterize an ILD.

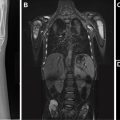
Another crucial consideration is normal growth and development. Progression in the severity or extent of imaging features in adult ILD is taken as evidence of progression of the causative disease process. However, in childhood, it is quite feasible that a single insult to the lung in infancy may be followed by substantial changes in the subsequent imaging appearance throughout childhood, not necessarily due to progression of disease, but possibly secondary to progression of lung growth around static disease. At present, very little longitudinal imaging data exist to characterize the expected imaging progression of ChILD, and consequently it is challenging to use structural imaging alone as the solitary means of assessing disease progression or treatment response in the setting of an interventional trial. Imaging appearance must be interpreted in tandem with clinical markers of disease severity ( Fig. 5 ).

A further relevant consideration when reporting CT findings in the setting of a ChILD is the importance of standardized language. The Fleischner Society glossary of terms, published in 2008, provides a set of internationally agreed upon terms and definitions that are essential to the classification of disease and the reproducibility of ILD imaging diagnosis. Varied use of terminology between adult and pediatric ILD imaging can become the source of considerable confusion. For example, the Fleischner Society definition of honeycombing includes “usually subpleural” “clustered cystic airspaces” and specifically mentions the need for caution in using the term because it is considered a specific imaging finding of UIP pattern fibrosis and its presence may impact patient care. One common pitfall to avoid is the misinterpretation of cysts seen in surfactant protein dysfunction as honeycombing, particularly by those less familiar with adult ILD practice; this has the potential to be misleading because the UIP pattern fibrosis seen in adult fibrotic lung disease is almost never encountered in pediatric ILD.
A retrospective review of biopsies of children with ChILD secondary to surfactant protein C mutations demonstrated 2 patterns of disease on histology: (1) alveolar macrophage accumulation with inflammatory cells and cholesterol clefts, type II pneumatocyte hyperplasia, and alveolar septal thickening by mesenchymal cells and lymphocytes and (2) respiratory bronchiole and duct dilatation with muscular hyperplasia. None demonstrated interstitial fibrosis, but cysts were demonstrated on CT in 40% at presentation and paraseptal emphysema in 13% with new cyst formation or increased cyst extent demonstrated on follow-up CT in most patients. Furthermore, although highly variable, the life expectancy and expected clinical course of children and adults with genetic disorders of surfactant function is generally different to the rapid decline seen in adults with progressive fibrosis as part of IPF. Therefore, the term honeycombing should be avoided in pediatric practice outside of those rare occasions in which multiple layers of subpleural cysts (adult pattern of UIP fibrosis) are demonstrated.
Ultrasonography
There is a growing trend, particularly among emergency and intensive care practitioners, of using ultrasonography as a primary modality for lung imaging, using the presence of differing artifacts (eg, ring-down or reverberation artifacts) at the pleural surface as a surrogate for the presence or absence of underlying lung disease. Although the technique may have some merits in low-resource settings, its inability to distinguish ILD from any other pathologic entity expanding the interstitium (eg, pulmonary edema or subsegmental atelectasis) renders it completely inadequate for the assessment of suspected ChILD ( Fig. 6 ).

There is, however, a clear role for ultrasonography in the assessment of extrapulmonary disease, which may in some cases prove vital to forming a specific diagnosis. Examples include (1) thymic and thyroid lesions that may be present in infantile Langerhans cell histiocytosis (LCH), (2) duodenal dilatation often seen in alveolar capillary dysplasia, (3) enlarged peripheral lymph nodes and hepatosplenomegaly characteristic of immunodeficiencies and hematologic malignancy, and (4) calcified liver lesions seen in Gaucher disease. In any patient presenting with ChILD with no specific diagnosis reached via CT, ultrasound examination of any relevant extrapulmonary viscera should be considered.
MR Imaging
Although pulmonary imaging by MR imaging has historically been limited by poor signal, high levels of susceptibility artifact at air-tissue interfaces, and the need for general anesthesia, there has been substantial advancement in the field of pulmonary MR imaging in recent years. Follow-up of structural lung abnormality by MR imaging is becoming more commonplace in the setting of specific lung diseases with gross structural pathology, particularly the bronchiectasis, marked bronchial wall thickening, and mucous plugging encountered in cystic fibrosis. The subtler structural abnormalities encountered in the setting of ChILD (eg, ground glass opacification and thin-walled pulmonary cysts) is far more difficult to appreciate with a conventional MR imaging scanner, but the recent advent of a low-field-strength (0.55 T) clinical MR unit with modern coil technology and sequences offers potential for the demonstration of structural ILD features in the near future. However, at present, the far higher spatial resolution of CT and the ability to perform ultrafast free breathing imaging even in newborns mean that MR imaging is unlikely to replace CT as the gold-standard imaging modality for the detection of ChILD for the foreseeable future. Instead, rather than structural assessment, arguably the most appealing use of MR imaging in the setting of ChILD research is the application of ventilation and pulmonary perfusion MR imaging techniques.
Methods of imaging ventilation via MR include the use of hyperpolarized noble gases, fluorinated gases, or even oxygen as contrast agents. Oxygen is weakly paramagnetic in its dissolved state and changes in signal result from local partial pressure of oxygen. Fluorinated and hyperpolarized gasses have the advantage of providing signal without being dissolved in fluid within the lungs, therefore providing a pure ventilation signal (oxygen-enhanced MR imaging outputs are a composite of ventilation and perfusion). Both oxygen and fluorinated gasses can be used to produce maps of ventilation and relative times for gas wash in and washout, akin to those measured via multiple breath washout lung function testing, likely of more use in airways disease than in ILD imaging.
An advantage of hyperpolarized noble gas imaging is the ability to quantify microscopic motion of a tracer gas within space. This imaging has been used to measure the size of individual airspaces with studies able to demonstrate catchup alveolarization far later in development than was previously thought possible in a cohort of teenagers with a history of premature birth. Furthermore, the Larmor frequency of xenon shifts as it changes in state from gaseous to dissolved form, and again as it binds to hemoglobin. This shift has allowed in vivo, spatially localized measurements of the process of gaseous diffusion at the alveolar membrane in healthy and diseased lungs, including adults with IPF. Although access to hyperpolarized gas MR imaging is currently limited to a handful of research centers worldwide, it is likely that the technique, alongside other methods of ventilation MR imaging, will have a substantial impact on the future of pulmonary imaging in bothadult and pediatric patient populations.
Pulmonary perfusion can also be imaged in several different ways. Contrast media can be injected with static or dynamic MR acquisitions demonstrating pulmonary arterial and parenchymal enhancement, often forming part of routine assessment in adults with pulmonary hypertension. Arterial spin labeling has also been used as a noncontrast method of pulmonary vascular imaging in children. Fourier decomposition imaging is another noncontrast MR technique for assessing ventilation and pulmonary perfusion in children by using the amplitude of signal changes at respiratory and pulse frequencies to form maps of pulmonary ventilation and perfusion without any injected or inhaled contrast media, requiring only 15 seconds per free breathing acquisition. ,
Last, MR lymphangiography is an increasingly well-established technique for assessing the pulmonary lymphatic system. Noncontrast techniques allow structural assessment of the central lymphatic system, and dynamic acquisitions following intranodal administration of contrast media are able to demonstrate the rate and direction of lymphatic flow ( Fig. 7 ). In fact, MR imaging is now capable of identifying pulmonary lymphatic pathology, such as lymphangiectasia and lymphangiomatosis, in fetal life with further characterization possible when contrast media can be delivered as a newborn.

Nuclear Medicine
A clear role has appeared for nuclear medicine in the monitoring of multisystem disease activity in adults with sarcoidosis. However, although some suggest a role in disease staging in pediatric LCH, there is currently little clinical utility of nuclear medicine studies in childhood ILD practice.
Interventional Radiology
Although transbronchial cryobiopsy has a possible role in adult ILD diagnosis, the size of the biopsy device precludes its use in ChILD diagnosis. Rarely, a specific ChILD may be diagnosable via image-guided biopsy of extrapulmonary tissues, for example, ultrasound-guided thyroid fine-needle aspiration in infantile LCH or peripheral lymph node biopsy in granulomatous lymphocytic ILD (GLILD).
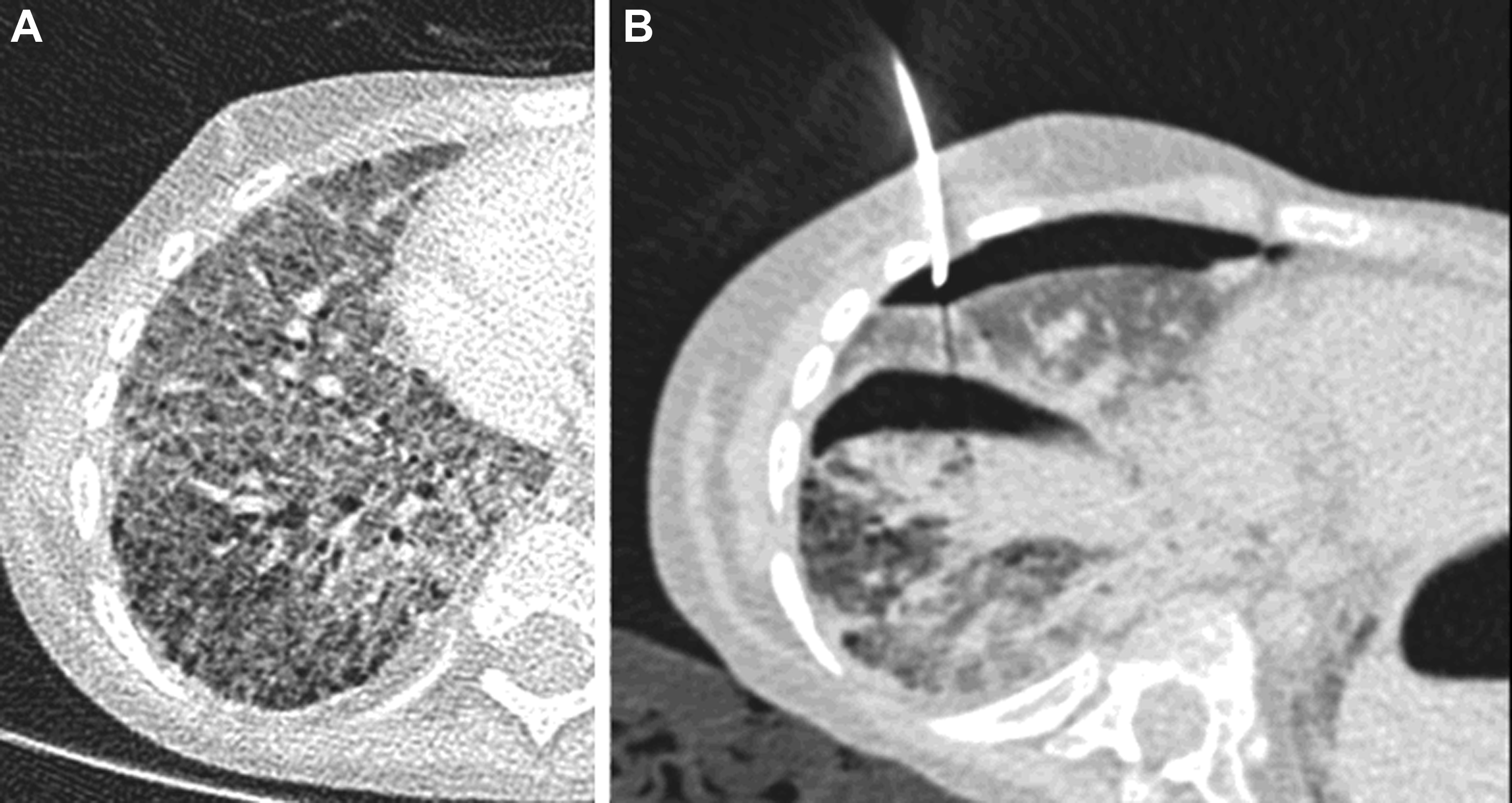
An additional role for interventional radiology in ChILD is pneumothorax drainage and management. Children with diffuse lung disease may suffer frequent pneumothoraces and may require interventional radiology placement of thoracic drains into small pneumothoraces made clinically important as a result of their extensive parenchymal disease. For these pediatric patients, a specific, very-low-dose protocol should be used because diagnostic image quality is not required ( Fig. 8 ).
Spectrum of pediatric interstitial lung disease
As discussed previously, the ChILD classification system mainly categorizes ILDs that occur in the pediatric population into 2 categories: disorders with onset in infancy and disorders not specific to infancy. In the following section, these 2 main categories of ChILD are systematically discussed alongside more recently described infant-onset ChILD entities not included within the most recent ATS classification.
Disorders with Onset in Infancy
The disorders with onset in infancy category is subdivided into (1) diffuse developmental disorders, (2) lung growth abnormalities, (3) specific disorders of undefined etiology, and (4) surfactant dysfunction disorders.
Diffuse developmental disorders
Diffuse developmental disorders are disorders of basic pulmonary development, characterized by the stage of developmental arrest. Typical clinical presentation varies with type, but is generally severe respiratory distress at or shortly after birth. The 2 main diffuse pulmonary developmental disorders are acinar dysplasia and alveolar capillary dysplasia with malalignment of the pulmonary veins (ACD-MPV).
Early developmental arrest results in acinar dysplasia, the complete lack of alveolar development, with affected infants surviving only hours postnatally. ACD-MPV is a misnomer because the pulmonary veins are not malaligned. The histopathologic appearance of transposition of the veins from their normal peripheral position in the interlobular septa to a centrilobular position is actually the result of dilated bronchial veins. There are well-known associations with FOXF1 mutations (up to 40% of reported ACD-MPV cases), and 80% are reported to have associated cardiac, renal, gastrointestinal, limb, or ocular malformations. The prognosis is generally bleak, with the affected patient often rapidly progressing to ECMO while the diagnosis is confirmed and one-way weaning. There are, however, reports of ACD-MPV presenting at several months of age, and prolonged survival has been reported in those with patchy disease. Despite the dramatic clinical presentation, CT imaging in ACD-MPV can be essentially normal, but a third of FOXF1 cases are reported to have concomitant pulmonary lymphangiectasia with resultant interlobular septal thickening and pleural effusions ( Fig. 9 ).