Abstract
Chondrodysplasia punctata refers to a group of skeletal dysplasias characterized primarily by punctate calcifications in cartilage (calcific stippling). These disorders are associated with short limb dwarfism, spinal abnormalities, facial dysmorphisms, joint contractures, skin lesions, and occasionally cardiac malformations. Ultrasound imaging shows premature calcifications in the joints and shortening of the appendicular skeleton. There are multiple types of chondrodysplasia punctata, with extensive locus and allelic heterogeneity.
Keywords
chondrodysplasia punctata, Conradi-Hünermann, peroxisome, stippling
Introduction
Chondrodysplasia punctata refers to a group of skeletal dysplasias that are characterized primarily by punctate calcifications in cartilage (calcific stippling). These disorders are characterized by short-limb dwarfism, spinal abnormalities, facial dysmorphisms, joint contractures, skin lesions, and occasionally cardiac malformations. There are multiple types of chondrodysplasia punctata, and there is extensive locus and allelic heterogeneity.
Disorder
Definition
The term chondrodysplasia punctata describes a group of osteochondrodysplasias characterized by punctate calcifications in cartilage, depending on the type and genetic alterations, and other organ system abnormalities.
Prevalence and Epidemiology
The heterogeneous bone dysplasias comprising chondrodysplasia punctata occur with an incidence of approximately 1 : 100,000 live births. The exact incidence for the specific types is unknown.
Etiology, Pathophysiology, and Embryology
There are six main types of chondrodysplasia punctata:
- •
rhizomelic form types 1, 2, and 3 (autosomal recessive)
- •
Conradi-Hünermann syndrome 2 (X-linked dominant) (CDPX2)
- •
brachytelephalangic (X-linked recessive) (CDXP1)
- •
autosomal dominant type (autosomal dominant)
Conradi-Hünermann syndrome is an X-linked dominant form that is due to a deficiency of delta(8)-delta(7) sterol isomerase emopamil-binding protein. This form is probably lethal in the hemizygous state in affected males. Affected females have elevated serum levels of 8(9)-cholesterol and 8-dehydrocholesterol. The brachytelephalangic type of chondrodysplasia punctata (CDXP1) results from mutations in the gene that encodes arylsulfatase E. In the autosomal dominant type of chondrodysplasia punctata and the very similar tibia-metacarpal type, the molecular defect has not yet been defined.
Manifestations of Disease
Clinical Presentation
The clinical characteristics of rhizomelic chondrodysplasia punctata are usually severe and include microcephaly, frontal bossing, micrognathia, congenital cataracts, cleft palate, respiratory insufficiency, calcific stippling in cartilage, kyphoscoliosis, symmetric shortening of the proximal long bones (rhizomelia), joint contractures, ichthyosis, alopecia, seizures, and cortical atrophy. Affected children usually die by 2 years of age.
Conradi-Hünermann syndrome (CDXP2) is an X-linked dominant type disorder characterized by failure to thrive in early infancy, mild to moderate growth deficiency, abnormal ears, deafness, cataracts, flattened midface, calcific stippling, scoliosis, hemivertebrae, asymmetric limb shortening, ichthyosis, coarse sparse hair, and occasional mental retardation. The brachytelephalangic type is an X-linked recessive disorder, and its manifestation is similar to other forms of chondrodysplasia punctata. Affected individuals have short stature, deafness, cataracts, facial dysmorphisms, calcific stippling in cartilage, distal phalangeal hypoplasia, and developmental delay. The autosomal dominant type of chondrodysplasia punctata and the similar tibia-metacarpal type are distinguished by dominant inheritance. Both manifest with short limbs; fine punctata are usually confined to the spine in the dominant form, and in the tibia-metacarpal type there is more discrete calcific stippling, particularly in the hands and vertebrae, short tibiae, and shortened second and third or third and fourth metacarpals.
Imaging Technique and Findings
Ultrasound.
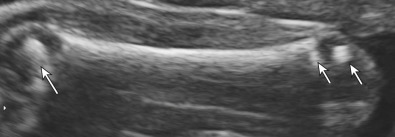
Ultrasound (US) findings can help identify the type of chondrodysplasia punctata. In all forms, there is evidence of cartilaginous stippling usually in the proximal humeri and femora, the distal portions of the femora, and the calcaneus. In many cases, this stippling leads to significant shortening of the rhizomelic portions of the bones. Depending on the severity, stippling may be apparent in the second trimester ( Figs. 48.1 and 48.2 ).