Abstract
The choroid plexus is a vascular convolute consisting of epithelial cells (microglia), fenestrated blood vessels, and stroma within the ventricular system of the brain. It is the main source of cerebrospinal fluid (CSF).
Choroid plexus cysts (CPCs) appear as intracranial fluid-filled lesions within the developing choroid plexus of the lateral ventricles, with a prevalence from 0.18% to 3.6%. They are benign, transient findings. The typical ultrasound (US) finding is a round hypoechogenic cyst in the choroid plexus. These are of variable diameter, ranging from 3 mm to 20 mm with a thick echogenic wall. CPCs are typically seen during the second-trimester screening examination. Cysts decrease rapidly in size and mostly disappear before birth. CPCs are associated with an increased likelihood of trisomy 18. When isolated CPCs are detected, the prior risk (maternal age or prior risk assessment) should be the major factor in deciding whether to offer fetal karyotyping. Parents should be reassured about the favorable outcome in CPC. If associated anomalies are seen, a fetal karyotyping should be offered.
Choroid plexus papillomas (CPPs) are extremely rare (0.5%–3% of intracranial tumors in children) and usually histologically benign fetal intracranial tumors, arising from the choroid plexus, and classified as primary tumor grade I. The typical US finding is a hyperechoic hypervascularized intraventricular mass. The tumor causes ventriculomegaly or hydrocephaly in approximately 85% of cases. A planned cesarean section is the delivery mode of choice in severe hydrocephaly. Affected children typically become symptomatic during the first year of life with signs of increased intracranial pressure. CPPs can be cured by surgical resection with favorable outcomes (5-year survival rate of up to 100%).
Keywords
central nervous system, choroid plexus, choroid plexus cysts, choroid plexus papilloma, intracranial tumors
Introduction
The choroid plexus is a vascular convolute, consisting of epithelial cells (a type of microglia), fenestrated blood vessels, and stroma, which lie in the ventricular system of the brain. It is the main source of cerebrospinal fluid (CSF) and actively regulates the constituents in CSF. The choroid plexus epithelial cells and the tanycytes, special ependymal cells located in the floor of the third ventricle, constitute the blood-CSF-brain barrier that separates blood and CSF compartments. The choroid plexus begins to differentiate as a bulge of the pia mater into the ventricular ependyma in week 6 of gestation, shortly after closure of the neural tube. The choroid plexus is derived from an invagination of the neuroepithelium and is located within the brain substance, but in continuity with the meninges. It first appears on the roof of the fourth ventricle, then in the lateral ventricles, and finally in the third ventricle. The two main components of the choroid plexus are the epithelium derived from the neural tube epithelium and mesenchyme derived from the meninges.
Choroid plexus cysts (CPCs) form most likely due to aberrant growth during the ninth week of gestation of either neuroepithelium or angiomatous, interconnecting thin-walled capillaries within the matrix of the immature choroid plexus. CPCs appear as fluid-filled areas in the choroid plexus and are the most common congenital abnormalities of the choroid plexus. They are easily detectable on prenatal ultrasound (US).
The choroid plexus can also be affected by acquired disease processes, such as primary or secondary neoplasms, infections, and hemorrhage. Intracranial tumors arising from the choroid plexus such as choroid plexus papillomas (CPPs) are extremely rare congenital anomalies.
Disease
Definition
Choroid plexus cysts (CPCs) are most frequently transient and benign, often round and sonolucent spaces within the developing choroid plexus of the lateral ventricles. They were first described by Chudleigh et al. and were found to be associated with aneuploidy, especially trisomy 18.
Choroid plexus papilloma (CPP) is a very rare, usually benign, fetal intracranial tumor. According to the World Health Organization classification of tumors of the nervous system, this more differentiated tumor is classified as a primary tumor of the choroid plexus grade I, as distinguished from atypical choroid plexus papilloma (grade II) and choroid plexus carcinoma (grade III).
Prevalence and Epidemiology
CPCs are frequently detected during the second-trimester US screening examination. The reported prevalence of fetal CPC ranges from 0.18% to 3.6%, independent of fetal sex.
CPPs represent 0.5% to 3% of intracranial tumors in children and 5% to 20% of all perinatal brain tumors. They are the most common brain tumors in the first year of life (up to 20%). Postnatally there is a male predilection, but a higher incidence in girls with Aicardi syndrome.
Etiology and Pathophysiology
CPCs are thought to originate from neuroepithelial folds of the choroid plexus filling with CSF and debris. The collection of fluid is surrounded by choroid plexus tissue. At 6 weeks’ gestation, the onset of the development of choroid plexus, a bulge from the medial wall of the lateral ventricle, becomes covered with pseudostratified epithelium, and then lobulated with villi. The cells change from cuboid to columnar epithelium. The choroid plexus produces CSF beginning from ninth week of gestation, leading to expansion of the ventricular system. As the villi grow and become entangled, a cystic space is formed, which traps CSF. Most villi form at 13 to 18 weeks’ gestation. Fluid accumulates and results in the formation of CPCs, which can be detected by prenatal US. Usually, the CSF is released and the cysts resolve as the stroma decreases with increasing gestational age, mostly between 26 to 28 weeks’ gestation.
Choroid plexus and CPCs differ in vascularity. Kraus and Jirasek showed typical wavy longitudinal nonbranching capillary loops under the surface epithelium of the choroid plexus. In contrast, there is a net of interconnecting angiomatous irregular capillaries (capillary plexus) with variable volumes in the walls of CPCs.
CPCs are typically transient and benign findings. Without associated anomalies they should be considered normal variants. CPCs in low-risk populations confer a 1% aneuploidy risk, mostly for trisomy 18, but occasionally also for trisomy 21 and triploidy. CPCs are found in 30% to 53% of fetuses with trisomy 18 in the second trimester. In trisomy 18, CPCs are associated with other sonographic anomalies in the majority (80%–90%) of these cases, but in 11% of pregnancies with trisomy 18 CPCs, they were the only detected sign. They have no known association with other adverse outcomes when the karyotype is normal. Long-term outcome studies have shown that the prenatal finding of isolated CPC, in the absence of aneuploidy, was not associated with altered fetal growth and development, delayed infant and early childhood development, or significant neurocognitive or neurobehavioral delays. Even in a case series of fetal ventriculomegaly secondary to large CPCs, in all cases the lateral ventricles became normal or decreased in size during pregnancy, resulting in normal growth and development on long-term follow-up (4–6 years).
No other known fetal anomalies are linked to CPCs. All studies so far have shown that the presence of associated anomalies, but not of CPCs per se, are the most important predictor of a fetal chromosomal defect, followed by increased maternal age.
When isolated CPCs are found in a low-risk population, the likelihood of trisomy 18 increases 3.5-fold to 9-fold when assessing only this single marker. In a metaanalysis of 13 prospective studies involving 1346 second-trimester fetuses with isolated CPC, Yoder et al. found a significant association between isolated CPC and trisomy 18, with a likelihood ratio of 13.8. In association with a structural abnormality, the risk increase above baseline was almost 1800-fold. The risk of chromosomal abnormalities in the presence of isolated CPCs depends on the a priori risk. For example, based on maternal age alone, the risk changes from less than 1 : 2500 if the mother is 20 years old to about 1 : 50 if the maternal age is 45 years ( Table 33.1 ).
| Maternal Age (years) | RISK | ||
|---|---|---|---|
| Overall | Isolated Cyst | Plus One Other Abnormality | |
| 20–24 | 1 : 4500 | 1 : 2950 | 1 : 225 |
| 25–29 | 1 : 3600 | 1 : 2300 | 1 : 175 |
| 30–34 | 1 : 2000 | 1 : 1300 | 1 : 100 |
| 35–39 | 1 : 750 | 1 : 470 | 1 : 35 |
| 40–44 | 1 : 400 | 1 : 100 | 1 : 10 |
Other factors, such as serum analyte screening, can alter the calculated a priori risk. Gupta used data from 200,000 US examinations to calculate aneuploidy risk in pregnancies with fetal CPC. In women who had multiple-marker screening tests indicating no increased risk, who had otherwise normal-appearing fetuses, and who were younger than 32 years, isolated CPCs did not increase the risk of trisomy 18 sufficiently to indicate fetal karyotyping. From a metaanalysis of eight prospective trials published between 1990 and 2000, Demasio et al. concluded that there is no evidence that detection of isolated CPC in women younger than 35 years increases the risk of trisomy 18. CPCs were suggested as a possible minor marker of trisomy 21, but the prevalence of CPCs in fetuses with trisomy 21 and in the general population is the same (1.4%), so they should not be considered to increase the risk of trisomy 21.
CPP is a polygenetic disorder. Different genetic loci have been implicated, and chromosomal alterations such as +7q (65%), +5q (62%), +7p (59%), and +5p (56%) have been reported. Different pathways in tumorigenesis of CPPs are active, such as platelet-derived growth factor receptor (PDGFR), the tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) pathways (CASP8, TFRSF10C, and TFRSF10D), Notch3 signaling, and the transcription factor TWIST1.
CPP is usually a benign tumor of ependymal origin, arising from the neuroepithelial cells of the choroid plexus, most commonly at the level of the atria of the lateral ventricles (70%–80%) but also from the third ventricle (14%), fourth ventricle (7%), and interventricular foramen.
CPP histologically resembles the nonneoplastic choroid plexus, with highly differentiated cuboidal epithelial cells based on papillary fibrovascular tissue, without signs of increased mitotic activity, nuclear atypia, or altered vascularization, but higher cellular density. These tumors are classified as choroid plexus papilloma World Health Organization grade I. The noninvasive and slow-growing tumor blocks the physiologic drainage of the CSF in the subarachnoid cisterns and secretes additional CSF, causing ventriculomergaly.
Malignant transformation of CPPs is rare (5%–10%). The 5-year survival rate after surgery of CPP has improved from 50% in early studies to approximately 100% in more recent reports, due to advances in imaging, surgical approaches, and quality of intensive care. There is no established association between fetal and neonatal tumors and chromosomal aberrations, other than in two syndromes, Aicardi-Goutières syndrome and hypomelanosis of Ito.
Manifestations of Disease
Clinical Presentation
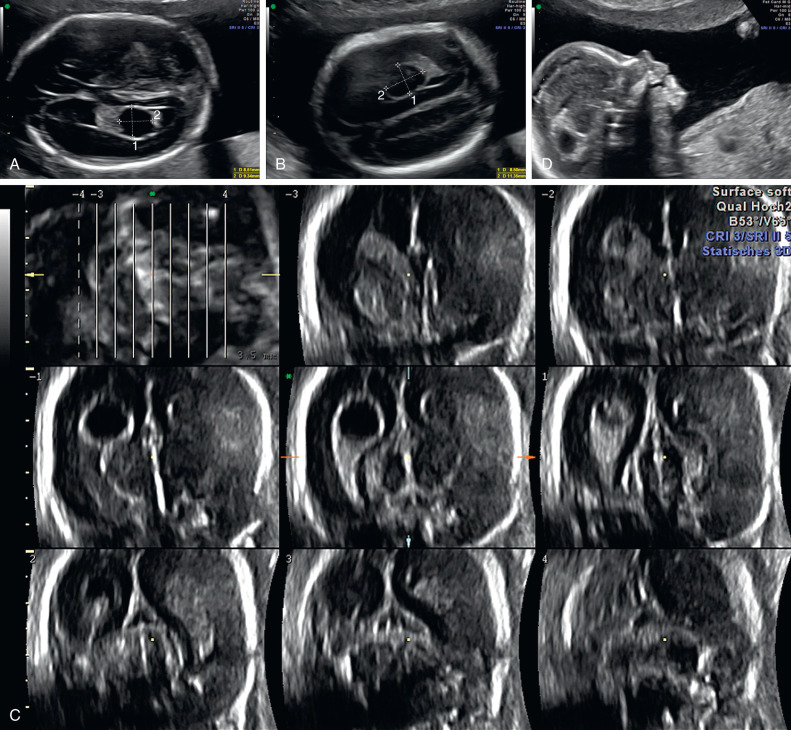
CPCs are single or multiple round hypoechogenic cystic structures usually in the body and atrium of the lateral ventricles, with diameters ranging from 3 mm to 20 mm. CPCs may be unilateral, bilateral, or multiple. Their appearance and laterality have no clinical relevance. Their cystic structure appears simple or septated; 95% of CPCs are isolated. The cysts do not cause any local damage or effect. Fetal CPCs may be detected by US as early as 11 to 12 weeks’ gestation. They resolve at the end of the second trimester in more than 95% of cases.
The prenatal diagnosis of CPP relies on the demonstration of an echogenic solid intraventricular mass at the level of the choroid plexus in the presence of ventriculomegaly, the main presenting finding in the fetus. Ventriculomegaly is present in 85% to 87% of affected cases, but correlates poorly with the size and pathologic features of the tumor. Affected children typically become symptomatic during the first year of life with signs of increased intracranial pressure. They usually experience headaches, nausea and vomiting, focal neurologic impairments, macrocephaly, and epileptic seizures. In rare cases, CPP can be a cause of intracranial hemorrhages.
Imaging Technique and Findings
Ultrasound.
The transventricular view is the first choice to visualize the normal choroid plexus. The plane lies superior and parallel to the transthalamic biparietal diameter plane. Before 16 weeks’ gestation, the choroid plexus fills most of the volume of the lateral ventricles with its typical symmetric butterfly appearance. The normal choroid plexus occupies the lateral ventricles from side to side.
CPCs can occur throughout the ventricular system, but they are typically found at the level of the atrium of the lateral ventricles. They are located in the body of the plexus, and may protrude into the ventricular cavity. They appear as sonolucent (black) spaces within the choroid plexus. CPCs show echogenic, well-defined walls and, occasionally, internal septations ( Figs. 33.1–33.3 and Table 33.2 ).