Melanoma stage
Description
Treatment options
0
The tumor confined to epidermis (melanoma in situ)
Surgical excision
IA
Tumor less than 1 mm thick without ulceration
Surgical wide excision
May consider SLNBa
IB
Tumor less than 1 mm with ulceration
Surgical wide excision
Consider SLNB
Or
1–2 mm without ulceration
IIA
Tumor 1–2 mm with ulceration
Surgical wide excision with SLNB
Or
2–4 mm without ulceration
IIB
Tumor 2–4 mm with ulceration
Surgical wide excision with SLNB
Or
>4 mm without ulceration
IIC
Tumor >4 mm with ulceration
Surgical wide excision with SLNB
IIIA
Tumor of any thickness with or without ulceration
Surgical wide excision and lymphadenectomy
IIIB
IIIC
Lymph nodes are involvedb
Consider adjuvant treatments, either on a clinical trial, with immunotherapy, or with radiation
IV
Metastatic
See text
M1a: metastases to skin, subcutaneous tissue, or distant lymph nodes – normal LDH level
M1b: metastases to lungs – normal LDH level
M1c: metastases to other organs, or any site with elevated LDH level
The mainstay of treatment for early melanoma is surgery, which helps staging patients and has a curative intent. Definitive surgery includes a wide excision with or without sentinel lymph node biopsy (SLNB). The role of SLNB on overall survival is unclear. The NCCN guidelines recommend a wide excision as category 1 evidence, but the SLN is only a category 2B and should be discussed and advocated for lesions thicker than 0.75 mm (Stage 1A) [5]. Sentinel lymph node biopsy is preferred over observation because it provides staging and prognostic information on the risk of stage upgrade with increasing Breslow [6]. The incidence of sentinel node micrometastases is 15–20 % in patients with intermediate thickness primary melanoma (1.2–3.5 mm). High risk features for positive sentinel lymph node are high mitotic rate, ulceration and lymph vascular invasion [5]. Patients with lymph node metastases should undergo lymphadenectomy which improves prognosis and survival rate and be offered adjuvant immunotherapy on a clinical trial or with interferon. Patients with metastatic disease need systemic therapy. In the last 5 years, there has been substantial development in the treatment of advanced melanoma. New targeted therapies and immunotherapies are benefiting a subset of patients who derive a longer survival [2]. Therapeutic options include targeted therapies, immune-based treatments, chemotherapy, or a combination thereof.
28.2 Molecular Signaling Pathways
The mitogen activated protein kinase (MAPK) pathway is activated in the majority of melanomas, through the neuroblastoma RAS viral oncogene homolog (NRAS) (15–20 %) or the v-raf murine sarcoma viral oncogene homolog B1 (BRAF) (40–50 %). NRAS and BRAF are components of the MAPK pathway, also called RAS-RAF-MEK-ERK signal transduction pathway (Fig. 28.1) [7]. Under physiological conditions the MAPK pathway transmits extracellular signals to the nucleus which leads to the expression of genes that drive cell proliferation, differentiation, and survival [8, 9]. The MAPK pathway is a critical component of oncogenic RAS signaling. In normal cells, the most important downstream mediators through this pathway are BRAF found in the testes, some hematopoietic precursors, and some brain cells, and CRAF which is essential to the daily function of most other cells. Both are serine/threonine kinases. The RAF proteins are major mediators of this pathway and signal through phosphorylation and activation of downstream kinases. RAF homodimerization or heterodimerization interacts with MEK and initiates its phosphorylation that leads to the phosphorylation and activation of ERK (also called MAPK), its substrate.
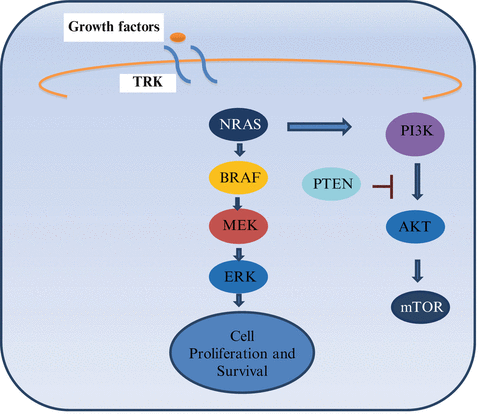
Fig. 28.1
MAPK pathway. TRK Tyrosine Kinase. When a ligand binds to a receptor on the cell surface it stimulates the activity of RAS. There are three isoforms of RAS: HRAS, KRAS and NRAS (RAS is the most commonly mutated oncogene in human malignancies). NRAS is commonly mutated in melanomas and can signal through MAPK and non MAPK pathways (PI3K pathways) [7, 8]
The activation of ERK leads to pro-growth signals that alter gene transcription. CRAF can have oncogenic effects through MEK independent pathways leading to nuclear factor kappa B (NF-kB) activation and inhibition of critical regulators of apoptosis (ASK-1 and MST-2). Activated BRAF has no other substrates other than MEK [8, 9].
There are more than 65 BRAF mutations reported in the literature. BRAF mutations occur most frequently in exon 15 at codon 600 (V600). The most common is BRAF V600E, comprising 90 % of all BRAF mutations. There are several substitutions that have been documented including valine by glutamic acid (V600E, 75 %), valine by lysine (V600K, 10–30 %), valine by arginine (V600R, 1–7 %) and lysine by glutamic acid (K601E, 1–4 %). Several characteristics are attributable to different BRAF mutations, as described in Tables 28.2 and 28.3 [7, 10]. Other pathway interferences by mutated BRAF include activation of NF-kB and others. For example, BRAF mutation (BRAF V600E) is also associated with activation of the mammalian target of rapamycin (mTOR) pathway. Activated ERK inhibits the tumor suppressor LKB1, a serine/threonine protein kinase mutated in autosomal dominantly inherited Peutz-Jeghers syndrome, a disease characterized by increased risk of benign and malignant tumors in multiple tissues. The LKB1 tumor suppressor negatively regulates mTOR signaling. ERBB 4 is activated in 19 % of melanomas, which leads to the activation of the PI3K pathway. This pathway involves PTEN and AKT, as described in Fig. 28.1. Normally PTEN is a tumor suppressor protein that negatively regulates the PI3K pathway, but when it is mutated it can activate the PI3k pathway by increasing expression of AKT. Selective activation of AKT (a downstream factor) is seen in 53 % of primary and 67 % of metastatic melanomas. PTEN mutation or deletion has been reported in up to 30 % of melanomas and can occur concurrently with BRAF, but not NRAS mutations [11].
Table 28.2
Mutations and their association with clinical presentations in melanoma
Mutations | Frequency | Clinical associations | Prognosis |
|---|---|---|---|
NRAS | 15–20 % | Skin with chronic or intermittent sun exposure | Worse prognosis in the metastatic setting |
BRAF | 40–50 % | Skin without chronic sun exposure | Worse prognosis in the metastatic setting |
c-KIT expression | 2–40 %a | Acral and mucosal melanomas | Unknown |
NF1, H–RAS, MAP2K1, MAP2K2, ERK phosphorylation, GNAQ and GNA 11 | Rare | GNAQ and GNA 11 seen in uveal melanoma | Unknown |
28.3 BRAF Testing
Testing for a BRAF mutation involves the extraction of genomic DNA from the tumor sample and a real time polymerase chain reaction (PCR) assay that detects both wild type and mutant BRAF. The Food and Drug Administration (FDA) has approved two tests cobas 4800 BRAF V600 Mutation Test (Roche Molecular Systems Inc., Pleasanton, CA, U.S.A.) and THxID®-BRAF KIT [12]. The Cobas 4800 test can identify 96 % of mutations across all specimen types with 5 % mutant alleles at a DNA input of 125 ng, an amount readily obtained from one 5 μm section of formalin-fixed paraffin-embedded tissue. The test can also identify V600K and V600D mutations, although the limit of detection is lower than that for V600E. Eighteen percent mutant alleles in a specimen are required for detection [13]. Other testing methods reported in the literature but not readily available in all institutions, include immunohistochemistry, pyrosequencing and next generation sequencing [12]. In our institution, BRAF testing is a send-out test and usually takes around 14 days to be reported. We recommend that the reader familiarizes him/herself with the turnaround time at their institution or vendors.
28.4 Chemotherapy for Metastatic Melanoma
Dacarbazine is the standard chemotherapy option for metastatic melanoma and the only FDA approved cytotoxic drug. The response rate is about 15 % with a median overall survival of 6–8 months [13]. Complete responses are observed in 5 % of patients with a 2–6 % survival at 5 years [14].
Temozolomide is an oral prodrug of the active metabolite of dacarbazine. It has been used to treat advanced melanoma and crosses the blood brain barrier, a theoretical advantage for patients with brain metastases. In a phase III study that compared temozolomide with dacarbazine in patients with no brain metastases, the median survival time was 7.7 and 6.4 months, respectively (HR 1.18; 95 % Confidence Interval (CI), 0.92–1.52). The median PFS was longer in patients who receive temozolomide (1.9 months) compared to dacarbazine (1.5 months) (p 0.012). There was no difference in overall survival or overall response rate [15].
Current NCCN guidelines list the following agents as category 2B for systemic chemotherapy of melanoma: nab paclitaxel, dacarbazine or temozolomide, dacarbazine, cisplatin, and vinblastine (CVD) with or without interferon alpha, and carboplatin with paclitaxel [5]. Combination chemotherapy usually yields a 25 % response rate with no improvement in survival. Biochemotherapy combines interleukin and interferon with CVD. This combination failed to demonstrate a survival benefit despite higher response rates. Chemotherapy remains a good option for patients who have potentially resectable oligometastases, and who obtain a response to systemic treatment given as a neoadjuvant modality prior to surgery. Patients should be carefully selected for this multidisciplinary approach.
28.5 Targeted Therapies for Metastatic Melanoma
28.5.1 c-KIT Inhibitors
In a phase II open label trial of 28 patients with advanced unresectable melanoma bearing a c- KIT mutation, imatinib, at 400 mg twice a day, yielded an overall response rate of 16 % (95 % confidence interval, 2–30 %) with a median time to progression of 12 weeks and a median overall survival of 46 weeks. While these results demonstrate the targeted effects, better patient selection is needed to narrow the targets that imatinib affects. Further studies with c- KIT inhibitors are underway in melanoma [16].
28.5.2 BRAF and MEK Inhibitors
To date, the FDA has approved three BRAF inhibitors vemurafenib, dabrafenib and trametinib, along with the combination of dabrafenib with trametinib (Table 28.4).
Table 28.4
Major clinical trials of BRAF inhibition
Therapy | Median time to response | Median duration of response (months) | Confirmed response (%) | Median PFS (months) | Median OS | Brain metastases |
|---|---|---|---|---|---|---|
Vemurafenib | 1.4 months | 6.7 | 48 | 5.3–6.8 | 84 % surviving at 6 months | No |
Dabrafenib | 1.5 months | 5.5 | 52 | 5.1 | N/A | No |
Trametinib + dabrafenib | NA | 10.5 | 76 | 9.4 | 72 % at 12 months | Yes |
Cobimetinib + vemurafenib | NA | NR | 68 | 9.9 | 81 % at 9 months | Yes |
28.5.2.1 Sorafenib
The first BRAF inhibitor to be tested was sorafenib, however a double-blind, randomized, placebo-controlled phase III study failed to improve overall survival when given in combination with carboplatin and paclitaxel for chemotherapy-naïve patients with metastatic melanoma. The median overall survival was 11.3 months (95 % CI, 9.8–12.2 months) for carboplatin and paclitaxel and 11.1 months (95 % CI, 10.3–12.3 months) for carboplatin, paclitaxel and sorafenib; the difference in overall survival distribution was not statistically significant. The reason for sorafenib failure could be attributed to the fact that is a non-specific inhibitor and that the trial included an unselected population [23].
28.5.2.2 Vemurafenib
Several clinical trials have established the clinical efficacy of vemurafenib for BRAF V600E mutated melanoma. The dose of vemurafenib is 960 mg orally twice a day. The overall response rate is 53 %, with 6 % and 47 % of complete and partial responses, respectively. The median duration of response is 6.7 months, the median progression-free survival (PFS), 6.8 months (95 % CI, 5.6–8.1), and the median overall survival, 15.9 months (95 % CI, 11.6–18.3 months) [24, 25]. The phase 3 trial, BRIM-3 Study Group, eventually led to FDA approval. The study enrolled 675 previously untreated patients with BRAF V600E mutated melanoma who were randomized between vemurafenib and dacarbazine. At 6 months, the overall survival was 84 % (95 % CI, 78–89) in the vemurafenib group and 64 % (95 % CI, 56–73) in the dacarbazine group. The study allowed a crossover from dacarbazine to vemurafenib. Vemurafenib was associated with a relative reduction of 63 % in the risk of death, compared to dacarbazine (p < 0.001). However, when the melanoma recurs, the prognosis is terrible. About 50 % of patients died of disease progression within 28 days of the last vemurafenib dose. Patients who progress after BRAF inhibitors have rapid clinical deterioration [2]. The most common adverse events included grade 1 and 2 photosensitivity, fatigue, alopecia, arthralgia, rash, serositis, keratoacanthoma and squamous cell carcinoma, and nausea and diarrhea. Squamous cell carcinoma was diagnosed in 18–26 % of patients [2, 25]. These skin cancers develop secondary to a paradoxical activation of the MAPK pathway and proliferation of HRAS Q61L transformed keratinocytes. This creates a decreased latency and accelerated growth of cutaneous squamous cell carcinomas and keratoacanthomas. Vemurafenib is not a tumor promoter but has been shown to accelerate the growth of preexisting RAS-mutant subclinical lesions [26]. All patients should have a dermatology evaluation before starting treatment with vemurafenib and a skin screening every 2 months afterwards. They should be aware of new lesions and report them to their oncologist. Before starting therapy it is recommended to perform an electrocardiogram to monitor for QT prolongation, to consult an ophthalmologist for a baseline eye exam, and to protect skin with regular sunscreen.
28.5.2.3 Dabrafenib
Dabrafenib is a selective inhibitor of mutant BRAF kinase. The first phase 1 study enrolled 184 patients including 156 patients with melanoma with or without asymptomatic brain metastases. The median PFS was 5.5 months in patients without brain metastasis and 4.2 months in patients with brain metastases. The dose is 150 mg orally twice a day. The phase 2 study enrolled 92 melanoma patients with histologically confirmed BRAF mutations (76 with BRAF V600E and 16 with BRAF V600K mutations). A 59 % response rate was seen in patients with BRAF V600E mutation, but only two patients with BRAF V600K mutation obtained a complete response. The median PFS was 6.3 months for BRAF V600E and 4.5 months for BRAF V600K. After a follow up of 11.9 months, the median overall survival was 13.1 and 12.9 months for BRAF V600E and BRAF V600K, respectively. The median time to response for BRAF V600E was 1.3 months [27]. The phase 3 study included 250 patients with stage IV or unresectable stage III BRAF V600E mutation positive melanoma randomly assigned to receive dabrafenib 150 mg orally twice a day or dacarbazine 1,000 mg/m2 intravenously every 3 weeks in a 3/1 ratio. The median PFS was 5.1 months for dabrafenib and 2.7 months for dacarbazine, with a hazard ratio of 0.30 (95 % CI 0.18–0.51; p < 0.0001) [17]. The most common adverse events were cutaneous squamous cell carcinoma, keratoacanthoma, fatigue, pyrexia, headache, nausea, and arthralgia. A panniculitis has also been described in patient obtaining remissions. The development of squamous cell carcinoma led to studies of the combination of BRAF inhibitors with MEK inhibitors to inhibit the squamous cell carcinoma pathway [27, 28].
28.5.2.4 Trametinib
Activated BRAF phosphorylates and activates MEK proteins (MEK1 and MEK2), which then activate downstream MAP kinases. Trametinib, a selective inhibitor of MEK1 and MEK2, is administered orally. The phase 3 study enrolled 322 patients with stage IIIC or IV cutaneous melanoma with a V600E (281 patients) or V600K BRAF mutations (40 patients). All patients were naïve to BRAF and/or MEK inhibition, or to ipilimumab. Patients with stable brain metastases were also allowed to enroll. Patients were randomized in a 2:1 ratio to 2 mg of trametinib once daily or chemotherapy consisting of either dacarbazine (1,000 mg/m2) or paclitaxel (175 mg/m2), every 3 weeks. The median PFS was 4.8 months in the trametinib group and 1.5 months in the chemotherapy group (HR, 0.45; 95 % CI, 0.33–0.63; P < 0.001). The 6-month overall survival rate was 81 % for trametinib and 67 % for chemotherapy. Crossover was allowed during this trial and 47 % of patients treated with chemotherapy crossed over to trametinib. The median duration of response was 5.5 months in the trametinib group. Adverse events of trametinib include rash, diarrhea, peripheral edema, fatigue, and dermatitis acneiform. There was a decrease in ejection fraction of 7 % in the trametinib group. There were no reports of cutaneous squamous cell carcinomas in patients receiving trametinib [18].
28.5.2.5 Cobimetinib
Cobimetinib is a potent selective MEK inhibitor, administered orally. The phase 3 study enrolled 495 patients with advanced stage IIIC or stage IV melanoma with a BRAF V600 mutation. They included patients with stable metastatic disease to the brain. Patients were randomized to vemurafenib + cobimetinib or placebo. The dose of vemurafenib was 960 mg BID and the dose of cobimetinib was 60 mg daily for 21 days and 7 days off. The median PFS was 9.9 months in the combination group and 6.2 months in the control group (HR for death or disease progression, 0.51; 95 % confidence interval [CI], 0.39–0.68; P < 0.001). The rate of CR or PR in the combination group was 68 %, and 45 % in the control group (P < 0.001). The interim analyses of overall survival showed 9-month survival rates of 81 % (95 % CI, 75–87) in the combination group and 73 % (95 % CI, 65–80) in the control group. Median duration of response was not reached in the combination group but was only 7.3 months in the vemurafenib and placebo arm. Adverse events in the combination group included central serous retinopathy, gastrointestinal events (diarrhea, nausea, or vomiting), photosensitivity, elevated aminotransferase levels, and an increased creatinine kinase level; most of them were grade 1 or 2. Most common grade 4 AE was elevation of creatinine kinase in the combination group (4 %), thought to be a class effect of MEK inhibition [19].
28.5.2.6 Combination of BRAF Inhibitors and MEK Inhibitors
In order to overcome resistance to BRAF inhibitors, several studies are underway to evaluate alternative combination of kinase inhibitors. Patients with BRAF V600 mutated metastatic melanoma were randomized to receive the combination of dabrafenib 150 mg orally daily and trametinib 1 or 2 mg, or dabrafenib monotherapy. The maximum tolerated doses for this combination were not reached in this study. The recommended phase 2 dose is the combination of dabrafenib 150 mg with trametinib 2 mg, which combines the recommended monotherapy dose for each agent. The median PFS of these 247 patients was 9.4 months for the combination and 5.8 months for single agent dabrafenib (HR, 0.39; 95 % CI, 0.25–0.62; p < 0.001). The overall response rate was 76 % for the combination group and 54 % for dabrafenib single agent (p = 0.03). Only 7 % of patients developed cutaneous squamous cell carcinomas when treated with the combination, but 19 % did with the monotherapy (p = 0.09). This combination was approved by the FDA in 2013.
In a preplanned interim overall survival analysis, the overall survival rate at 12 months was 72 % (95 % confidence interval [CI], 67–77) in the combination-therapy group and 65 % (95 % CI, 59–70) in the vemurafenib group (hazard ratio for death in the combination-therapy group, 0.69; 95 % CI, 0.53–0.89; P = 0.005). Median PFS was 11.4 months in the combination group and 7.3 months in the vemurafenib group (hazard ratio, 0.56; 95 % CI, 0.46–0.69; P < 0.001). The objective response rate in the combination group was 64 % and 51 % in the vemurafenib group (P < 0.001) [20, 21].
28.5.3 Mechanism of Resistance to BRAF Inhibitors
Tumor resistance develops in a median of 5–7 months. There are different mechanisms by which tumors develop resistance. The MAPK pathway dependent mechanism includes de novo mutations in NRAS (upstream) and MEK (downstream). Overexpression of mitogen-activated protein kinase kinase kinase 8 (MAP3K8) drives resistance to RAF inhibition in BRAF V600E cell lines. MAP3K8 activates ERK primarily through MEK-dependent mechanisms that do not require RAF signaling. Moreover, MAP3K8 expression is associated with de novo resistance in BRAF V600E cultured cell lines and acquired resistance in melanoma cells and tissue obtained from relapsing patients following treatment with MEK or RAF inhibitors [14]. Another MAPK independent pathway mechanism involves the overexpression or overactivation of PDGFR-β or IGF1R inducing oncogenic signaling through PI3K-AKT-mTOR pathway (Fig. 28.2) [30]. Resistance to the combination of dabrafenib and trametinib was tested by whole exome and whole transcriptome sequencing, on five patients with acquired resistance. Three patients had additional MAPK pathway alterations including a novel MEK2 mutation that conferred resistance to RAF/MEK inhibition in vitro [31]. Acquired resistance to these targeted therapies need to be further studied to determine alternative treatment strategies. These may include combination therapies, addition of downstream targeted therapies, and dosing adjustment, among others.
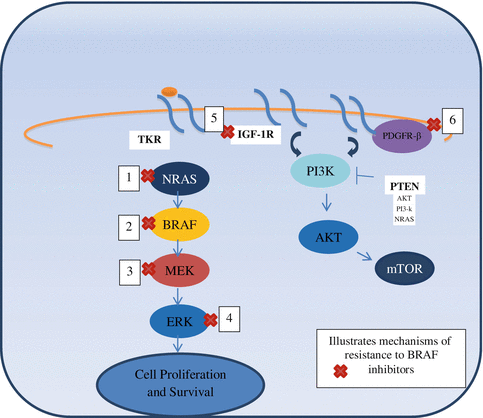
Fig. 28.2
Mechanisms of resistance to BRAF inhibitors [29]. 1. NRAS mutations. 2. BRAF V600E slice variant: creates a truncated form of BRAF mRNA and this mutated BRAF protein has enhanced interaction with RAS. This leads to dimer formation between the truncated, activated BRAF kinase and wild type RAF kinases. Once dimerized, BRAF inhibitors (such as Vemurafenib) can induce transactivation and then reactivation of MAPK pathway. 3. MEK–1 mutation. 4. BRAF inhibitors lead to decreased activation of ERK. There is decreased level of negative regulators which then leads to decreased suppression of RAS. RAS reactivates and then dimerizes and activates BRAF. 5. IGFR activation leads to non-MAPK pathway activation. 6. PDGFRβ activation leads to non-MAPK pathway activation
A phase I/II trial evaluated the combination of dabrafenib and trametinib after disease progression with a BRAF inhibitor. The ORR was 14 % (95 % CI, 7–24 %), and an additional 46 % of patients had stable disease 8 weeks; median PFS was 3.6 months. This regimen may be a therapeutic strategy in patients who had previously been exposed to single agent BRAF inhibitor for >6 months. In patients with rapid development of resistance, less than 6 months, derived no benefit on further combination therapy and had rapid progression [32].
28.6 Immunotherapy for Metastatic Melanoma
Melanoma is associated with immune-related phenomena, including spontaneous remission in the absence of active therapy or vitiligo. Rare patients who developed infections and fever have been found to have tumor regression [33]. About 16 % of patients with advanced melanoma respond to high-dose interleukin-2 (IL-2), a non-specific type of immunotherapy that activates T cells [34]. Cytotoxic T lymphocyte (CTL) activation requires antigen-specific recognition. Co-stimulatory and co-inhibitory signals are also required to orchestrate this process [35] (Fig. 28.3 and Table 28.8). Immunomodulation of co-inhibitory signals, including CTLA-4 and PD-1, have become pivotal targets for the treatment of melanoma. In the last 5 years, such new targeted immunotherapy drugs have revolutionized the treatment of advanced melanoma. Gradual understanding of immune-specialized cell interplay will lead to newer therapeutic approaches.
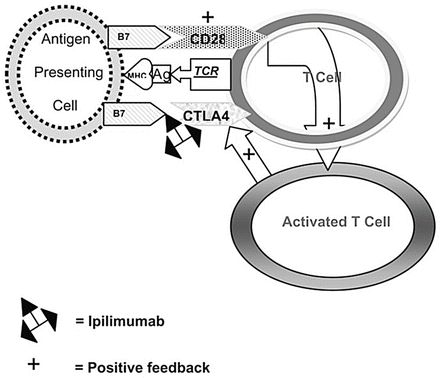
Fig. 28.3
T cell activation and mechanism of action of ipilimumab. When an antigen (Ag) is presented in the context of the major histocompatibility complex (MHC) to the T cell receptor (TCR), binding of B7 with CD28 occurs which activates the T cell. Slightly later, the activated T cell stimulates CTLA4 which also binds to B7 to down-regulate the T cell. Ipilimumab inactivates the binding of CTLA4 with B7, allowing the T cell to remain activated [36]
28.6.1 Evaluation of Response after Immunotherapy for Melanoma
Response Evaluation Criteria in Solid Tumors (RECIST) or WHO criteria are conventionally employed to evaluate the response to chemotherapy in solid tumors. Tumor response to immunotherapy has a different pattern. Tumor shrinkage induced by immunotherapy may be preceded by inflammatory changes, initially causing tumor swelling. New immune-related response criteria (irRC) have been proposed [49]. The irRC approach attempts to not separate index lesions from new lesions. Instead, irRC considers index lesions and new measurable lesions together to measure total tumor burden and defines immune-related complete response (irCR), immune-related partial response (irPR), and immune-related stable disease (irSD). As long as the total tumor burden is decreased to more than 50 %, progression of some lesions or the appearance of new lesions is acceptable to adjudicate partial response. In most clinical trials of immunotherapy in advanced melanoma, irRC are used with RECIST and/or WHO criteria in parallel or in tandem (Table 28.5).
Table 28.5
Comparison between WHO criteria and irRC
WHO | irRC | |
|---|---|---|
CR | Disappearance of all lesions in two consecutive observations not less than 4 weeks apart | Disappearance of all lesions in two consecutive observations not less than 4 weeks apart |
PR | ≥50 % decrease in SPD of all index lesions compared with baseline in two observations at least 4 weeks apart, in absence of new lesions or unequivocal progression of non-index lesions | ≥50 % decrease in tumor burden compared with baseline in two observations at least 4 weeks apart |
SD | 50 % decrease in SPD compared with baseline cannot be established nor 25 % increase compared with nadir, in absence of new lesions or unequivocal progression of non-index lesions | 50 % decrease in tumor burden compared with baseline cannot be established nor 25 % increase compared to nadir |
PD | At least 25 % increase of SPD compared with nadir (or unequivocal progression of non-index lesion) and/or appearance of any new lesions (at any single time point) | At least 25 % increase of tumor burden compared to nadir (at any single time point) in 2 consecutive observations at least 4 weeks apart |