Congenital Anomalies
UPPER URINARY TRACT ANOMALIES
Kidney
Anatomy
The kidneys are paired retroperitoneal structures that parallel the psoas muscle on either side of the lumbar spine. They are composed of a variable number of renal pyramids, each of which consists of a minor calyx and its associated ducts. The base of the pyramid is formed by its overlying renal cortex and the apex is the renal papilla, which projects into a minor calyx. The papillae contain the openings of the distal collecting ducts (of Bellini), which empty into the calyces. The calyx is the cup-shaped portion of the intrarenal collecting system and the rim of the cup is the fornix. Urine drains from each calyx into an infundibulum and then into the renal pelvis.
The kidney is divided into an outer cortex and an inner medulla by the arcuate artery at the base of each pyramid. Columns of cortical tissue sometimes descend between the medullary pyramids and are often referred to as a column of Bertin. The calyces, infundibula, and renal pelvis are referred to as the “intrarenal collecting system.” The renal sinus is the space around the collecting system and contains a variable amount of fat, along with branches of the renal artery, vein, and lymphatics.
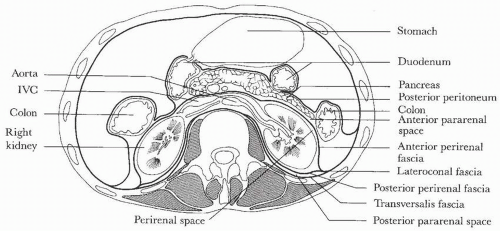 FIGURE 1.1. Retroperitoneal anatomy. (From Amis ES Jr, Newhouse JH. Essentials of Uroradiology. Boston, MA: Little, Brown & Co.; 1991:5, with permission.) |
The retroperitoneum is defined by fascial layers and divided into three compartments: the anterior pararenal space, the perirenal space, and the posterior pararenal space (Fig. 1.1). The anterior pararenal space contains the pancreas, the second through fourth portions of the duodenum, the ascending and descending colon, and the hepatic and splenic arteries. The perirenal space is defined by the anterior and posterior layers of Gerota fascia. These two layers may fuse in the midline or may extend across the midline without fusing, allowing communication between the two perirenal spaces. The kidney, adrenal gland, and proximal ureter are contained within Gerota fascia in the perirenal space. The posterior pararenal space contains only fat.
Anomalies of Position
Malrotation
Malrotation of a normally positioned kidney most commonly occurs as a result of failure of rotation of the kidney about its vertical axis. This results in an anterior position of the renal pelvis. It may be bilateral or unilateral, and in rare circumstances, overrotation of the kidney may result in a laterally facing renal pelvis. On computed tomography (CT), the renal pelvis will be seen projecting anteriorly (Fig. 1.2).
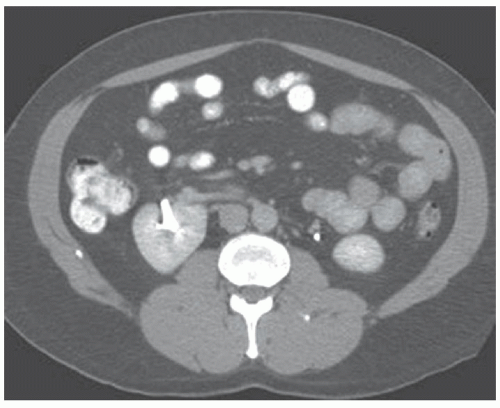 FIGURE 1.2. Malrotated kidney. On CT, the renal pelvis projects anteriorly. |
Renal Ectopy
As the fetal kidneys ascend from the pelvis, each kidney acquires blood supply from the neighboring vessels. The initial supply from the external and internal iliac vessels is lost during this process, and blood supply directly from the aorta is acquired around the 8th week of development. An abnormality in acquiring such blood supply may prevent cephalic migration from occurring and result in renal ectopy or an abnormal position of the kidney. The most common form of renal ectopy is a pelvic kidney in which the kidney is located in the true pelvis or anterior to the sacrum (sacral kidney). The reported incidence of renal ectopy varies between 1:500 and 1:1,200. The exact incidence is difficult to determine, because renal ectopy is often clinically silent. With any congenital anomaly, the likelihood of finding other anomalies in the same or other organ systems is increased. Therefore, it is not unusual to find pelvic kidneys associated with a pathologic process, such as hydronephrosis or vesicoureteral reflux. Clinical symptoms related to the presence of a pelvic kidney are usually due to one of its associated conditions, such as pain from obstruction or infection related to reflux.
The findings depend on the degree of renal function in the pelvic kidney and the presence of associated abnormalities. On ultrasound, the reniform mass will be found in the pelvis with a characteristic pattern of renal sinus echoes. On CT, a functioning mass of renal parenchyma can usually be identified (Fig. 1.3). Angiography is often employed before any contemplated surgical procedure on a pelvic kidney because of the highly variable nature of its blood supply.
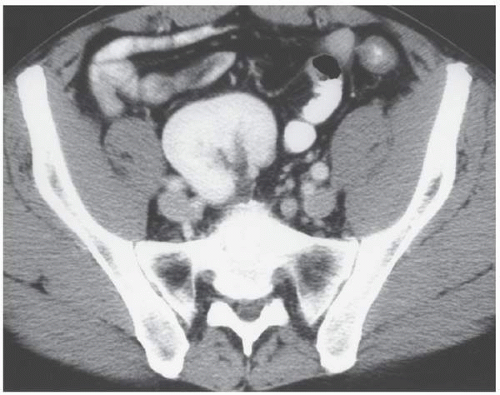 FIGURE 1.3. Pelvic kidney. A contrast-enhanced CT examination demonstrates the right kidney in the pelvis. |
Intrathoracic Kidney
This rare anomaly occurs when the kidney ascends to a position higher than the second lumbar vertebra. The kidney may reach an intrathoracic location through a posterior diaphragmatic aperture that may be congenital or acquired. The anomaly is more common in males and is more commonly found on the left side. The blood supply to an intrathoracic kidney generally arises from the abdominal aorta in its normal location. Congenital intrathoracic kidney occurs in 1:15,000 births and often is diagnosed by finding a posterior thoracic mass, which appears to arise from the diaphragm on a chest radiograph.
Anomalies of Number
Renal Agenesis
True renal agenesis is the complete congenital absence of renal tissue (Fig. 1.4). This condition is to be distinguished from acquired forms of agenesis, in which renal tissue develops but atrophies during development or during childhood because of an associated malfunction. Renal agenesis occurs in approximately 1 in 1,000 births. It is thought to occur as the result of failure of formation of the ureteral bud or because of an inherent deficiency of the metanephric blastema. In the latter case, partial development of the ureter may be present. In true agenesis, ipsilateral absence of the trigone and ureteral orifice will be found in the bladder on cystoscopy. No renal artery is present, and the colon occupies the renal fossa on the affected side. The ipsilateral adrenal gland is absent in 8% to 10% of cases. Compensatory hypertrophy of the contralateral kidney almost always accompanies renal agenesis and can also be found when the other kidney is compromised by any other process, is removed, or is functionally compromised. However, the older the patient at the time of renal damage, the less compensatory hypertrophy will occur.
Genital abnormalities may also be associated with unilateral renal agenesis and, when present, suggest an etiology that also affects the mesonephric duct. In males, such abnormalities include cysts or absence of the ipsilateral seminal vesicle, absence of the ipsilateral vas deferens, hypoplasia or agenesis of the testicle, and hypospadias.
In females, renal agenesis may be associated with a unicornuate or bicornuate uterus, absence or hypoplasia of the uterus, and
absence or aplasia of the vagina, the Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH syndrome). Often considered the male counterpart of the MRKH syndrome, Zinner syndrome includes unilateral renal agenesis, a cyst in the ipsilateral seminal vesicle, and obstruction of the ejaculatory duct. Men with Zinner syndrome are often diagnosed during adulthood because of infertility.
absence or aplasia of the vagina, the Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH syndrome). Often considered the male counterpart of the MRKH syndrome, Zinner syndrome includes unilateral renal agenesis, a cyst in the ipsilateral seminal vesicle, and obstruction of the ejaculatory duct. Men with Zinner syndrome are often diagnosed during adulthood because of infertility.
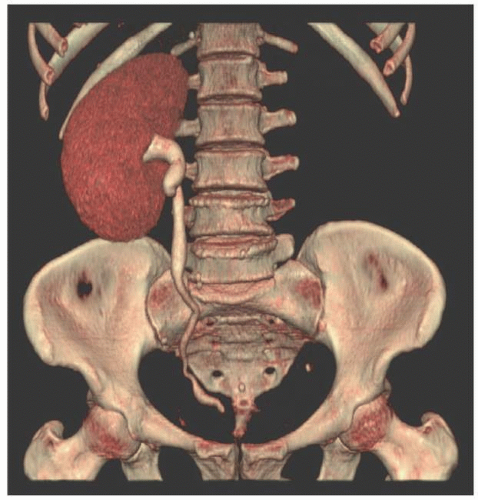 FIGURE 1.4. Renal agenesis. A volume-rendered image from a CT urogram shows a single right kidney to be present. (Courtesy of Seog Wan Ko, MD, Department of Radiology, Gwangju Christian Hospital, Gwangju, South Korea.) |
Bilateral renal agenesis is extremely rare and incompatible with life. Males are affected in three-fourths of the cases. Infants with this abnormality exhibit the characteristic features of Potter facies, which include low-set ears and a prominent palpebral fold.
Renal Hypoplasia and Dysplasia
Renal hypoplasia is an unusual renal anomaly in which the kidney is at least 50% smaller than normal and contains fewer than the normal number of calyces. The condition is usually unilateral, and the kidney functions normally for its size. Most unilaterally small kidneys are acquired because of chronic ischemia, reflux (chronic atrophic pyelonephritis), or long-standing obstruction (hydronephrotic atrophy). However, these conditions can be differentiated from hypoplasia by the fact that in reflux or obstruction, the calyces are abnormally cupped.
The Ask-Upmark kidney (see Chapter 10) was thought by some to be a variant of renal hypoplasia. The kidney is small, predominantly because of cortical loss in the upper pole associated with cortical indentations. However, most patients also have associated reflux and infection. The Ask-Upmark kidney is currently thought to be caused by scarring due to chronic pyelonephritis rather than a true congenital lesion.
Supernumerary Kidney
A supernumerary kidney is extremely rare. Cleavage of the metanephric blastema has been suggested as the cause for this abnormality. Most supernumerary kidneys are caudally placed and are hypoplastic. They may be connected to the ipsilateral dominant kidney either completely or by loose areolar connective tissue. A separate collecting system in the supernumerary kidney is generally present.
Anomalies of Form
Crossed Ectopy
Crossed ectopy is defined as a kidney located on the opposite side of the midline from its ureter. The crossed kidney usually lies below the normally situated kidney. In 90% of the cases, at least partial fusion between the kidneys is present (crossed-fused ectopy). In the remainder, two discrete kidneys on the same side are present (crossed-unfused ectopy). Other variations of crossed ectopy, including solitary crossed ectopy and bilateral crossed ectopy, have been described (Fig. 1.5). The anomaly is more common in males (2:1), and left-to-right ectopy is three times more common than right-to-left ectopy. Crossed-fused ectopy has been estimated to occur in approximately 1:1,000 births.
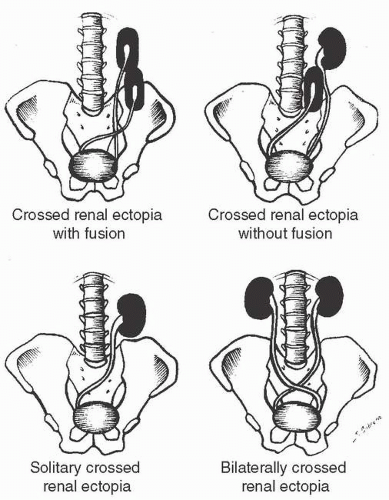 FIGURE 1.5. Schematic drawing showing the variations of crossed renal ectopy. |
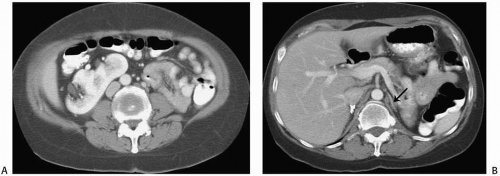 FIGURE 1.6. Crossed-fused renal ectopy. A: The left kidney lies in the right abdomen and is fused to the right kidney. B: The left adrenal gland (arrow) has a single limb. |
The anomaly is thought to result because of an abnormally situated umbilical artery that prevents normal cephalic migration from occurring; in such cases, the developing kidney takes the path of least resistance and crosses to the opposite side, where cephalic migration resumes. Others have postulated that the abnormality occurs when the ureteral bud crosses to the opposite side, where it induces nephron formation in the contralateral metanephric blastema.
In most cases of crossed renal ectopy, the ureters are not ectopic and cystoscopy reveals a normal trigone. The incidence of associated congenital anomalies is low. Symptoms of crossed renal ectopy are rare; however, these patients may present in adulthood with abdominal pain, pyuria, or urinary tract infection. A slightly higher incidence of urinary tract calculi associated with crossed renal ectopy is thought to be related to stasis.
The abnormality is readily detected on cross-sectional imaging studies (Fig. 1.6). On ultrasound, crossed-fused ectopy can be
identified by a characteristic anterior or posterior “notch” between the two kidneys. The blood supply to the crossed renal unit is generally anomalous; as with pelvic kidney, angiography is usually recommended before surgical procedures.
identified by a characteristic anterior or posterior “notch” between the two kidneys. The blood supply to the crossed renal unit is generally anomalous; as with pelvic kidney, angiography is usually recommended before surgical procedures.
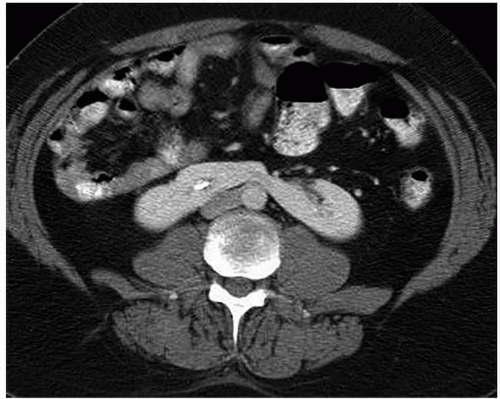 FIGURE 1.7. Horseshoe kidney. An enhanced CT demonstrates fusion of the two renal units. |
Horseshoe Kidney
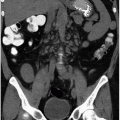
Horseshoe kidney is the most common renal anomaly, occurring in approximately 1:400 births. There is a 2:1 male predominance. The abnormality occurs when the kidneys are connected by an isthmus. The anomaly is thought to occur because an abnormal position of the umbilical artery results in disturbance of the normal pattern of cephalic migration. As a result, there is contact between the developing metanephric blastema on each side that leads to partial fusion.
The isthmus of a horseshoe kidney usually consists of a band of parenchymal tissue (Fig. 1.7) that has its own blood supply. In some cases, the band consists of only fibrous tissue. Usually, the band joins the lower poles of the kidney and prevents normal rotation from occurring, so that on each side, the renal pelvis is in an anterior position. Rarely, the band connects the upper poles rather than the lower poles. The band is usually located anterior to the aorta and inferior vena cava (IVC), but posterior to the inferior mesenteric artery, which is thought to prevent further cephalad migration from occurring. However, variations are common so that the position of the kidneys, their blood supply, their relation to the major vessels, and even the size of the kidney on either side are variable. Horseshoe kidney is commonly associated with ureteropelvic junction (UPJ) obstruction (Fig. 1.8). Many patients with horseshoe kidney remain asymptomatic throughout their lifetimes; in the remainder of patients, symptoms of obstruction, infection, or a renal calculus bring the abnormality to medical attention. A horseshoe kidney is more prone to injury with blunt abdominal trauma, presumably because it is relatively less protected in its more anterior position.
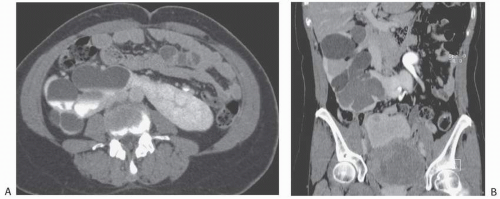 FIGURE 1.8. Horseshoe kidney with UPJ obstruction. A: Axial CT image reveals severe hydronephrosis of the right portion of the horseshoe kidney with only a thin rim of functioning parenchyma remaining. B: Coronal reformatted image. (Courtesy of Seog Wan Ko, MD, Department of Radiology, Gwangju Christian Hospital, Gwangju, South Korea.) |
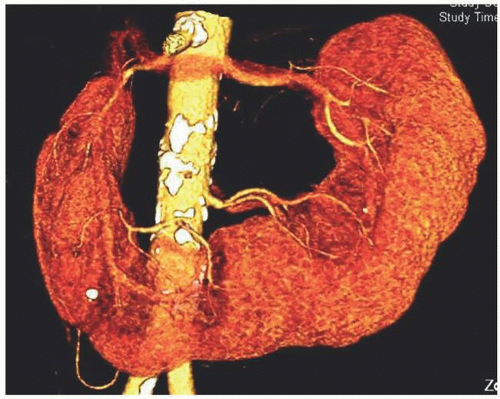 FIGURE 1.9. Horseshoe kidney. Reconstructed images from a CT urogram demonstrate multiple arteries supplying a horseshoe kidney. |
The blood supply to a horseshoe kidney is quite variable, most patients having multiple bilateral renal arteries (Fig. 1.9). The isthmus may be supplied by branches of the renal arteries or directly from the abdominal aorta, the inferior mesenteric artery, or the iliac arteries.
Imaging findings in patients with a horseshoe kidney include (1) an abnormal axis for each kidney with the lower poles more medially located than the upper poles, (2) the kidneys lie in a more caudad position, and (3) a bilateral malrotation with the renal pelves in an anterior position so that the lower calyces lie in a more medial position than the proximal ureter. On CT or MR, the isthmus of a horseshoe kidney is usually easily identified and may also be helpful in defining the relationship of the kidney to the major vessels.
Other Fusion Anomalies
Various other fusion abnormalities occur much less commonly than a horseshoe kidney or crossed renal ectopy. A doughnut kidney refers to two kidneys joined medially at their poles to form a ringlike renal mass. The lump, or pancake, kidney is a rare abnormality marked by extensive fusion between the two renal masses. The kidney is found in the midline or slightly to one side, generally no higher than the sacral promontory. The renal pelves are anterior and drain separate portions of the kidney. The ureters generally do not cross.
Renal Pelvis and Ureter
Unipapillary Kidney and Polycalycosis
The normal human intrarenal collecting system typically has 10 to 14 calyces. On rare occasions, a unipapillary kidney can be found in humans. It is more commonly found involving the left kidney and is usually associated with other significant anomalies, such as ipsilateral hypoplasia and frequent abnormalities of the contralateral kidney. Conversely, polycalycosis signifies an increased number of calyces; this is usually an isolated finding in an otherwise normal kidney.
Congenital Megacalyces
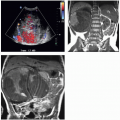
Many investigators believe that congenital megacalyces, also known as megacalycosis, is an acquired condition. The calyces are all symmetrically enlarged, although the renal pelvis and ureter are of normal size (Fig. 1.10). Furthermore, there is no history to suggest previous obstruction or reflux. Involvement is almost always unilateral, and the kidney is typically normal in size and function. Although megacalyces may be congenital, clinically silent obstruction or reflux in the distant past, perhaps even during fetal life, may be the cause. Another factor that favors an acquired etiology is that megacalycosis is typically seen in adults.
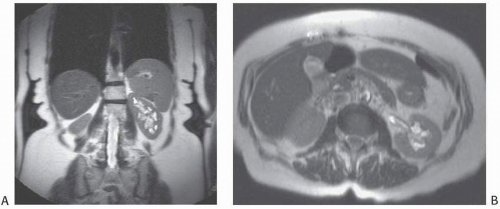 FIGURE 1.10. Congenital megacalyces. A: Coronal. B: Axial MR shows diffuse enlargement of calyces in the left kidney. The left renal pelvis is not dilated. |
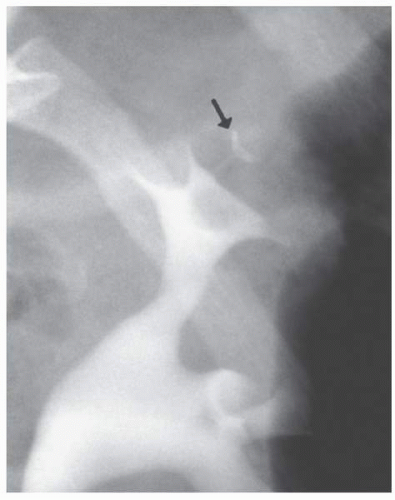 FIGURE 1.11. Microcalyx. A small, but otherwise normalappearing calyx (arrow) is seen in the upper pole. |
Microcalyx
A microcalyx resembles a normal calyx in every way but size (Fig. 1.11). It may arise from the renal pelvis or an infundibulum and ends at a papillary tip. The microcalyx is formed normally with a fornix and tubules draining into it.
Aberrant (Ectopic) Papilla
Most renal papilla empty into a minor calyx. In the renal polar regions, where compound calyces are common, several papillae may empty side by side into a major calyx. Rarely, a papilla may have an aberrant insertion into the collecting system and may present as a filling defect that must be differentiated from stones, tumors, or other pathologic processes. The aberrant papilla can protrude into virtually any part of the collecting system surrounded by renal parenchyma, including an infundibulum or the intrarenal portion of the renal pelvis. The resultant finding on excretory urography or retrograde pyelography is a smooth round or ovoid lucent defect in the contrast-filled collecting
system (Fig. 1.12). The smooth border and fixed location on a margin of the collecting system tend to differentiate the finding from a tumor or stone. Oblique views may show the extrinsic origin of the papilla.
system (Fig. 1.12). The smooth border and fixed location on a margin of the collecting system tend to differentiate the finding from a tumor or stone. Oblique views may show the extrinsic origin of the papilla.
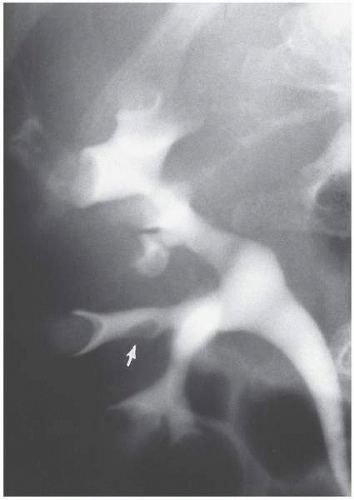 FIGURE 1.12. Ectopic papilla. A smooth ovoid filling defect (arrow) in an infundibulum is seen on an excretory urogram. |
Ureteropelvic Junction Obstruction and Congenital Megaureter
Although occurring at either end of the ureter, UPJ obstruction and congenital megaureter are discussed together because of their histologic and physiologic similarities. Both conditions are caused by a deficiency and derangement of the ureteric smooth muscle fibers with associated fibrosis, resulting in a failure of normal peristalsis in the affected segment and subsequent functional obstruction. Congenital obstruction of the UPJ is a common anomaly of the urinary tract. The disorder produces caliectasis and marked pelviectasis as a result of a functional narrowing of the UPJ.
In approximately 5% of cases, extrinsic compression of the UPJ by an aberrant renal artery results in a similar radiographic pattern. Congenital UPJ obstruction is the most common cause of an abdominal mass in a neonate. The disorder is being discovered increasingly in the antenatal period because of the almost routine use of obstetric ultrasound. The abnormality may be clinically silent until adulthood, when hematuria, flank pain, fever, or, rarely, hypertension causes the patient to seek medical attention. Cases in which presentation has been delayed into the sixth or seventh decade have been reported. In some patients, it is an incidental finding on studies performed for another indication. Males are affected more often than females by a 2:1 ratio, and the left side is more commonly affected than the right for unknown reasons. A familial tendency toward the disorder has also been reported.
In some patients, symptoms may be present only during sustained diuresis, a condition known as “beer-drinker’s hydronephrosis” or Dietl crisis. Such cases are attributed to a very mild UPJ obstruction, such that under conditions of normal urine flow rates, the UPJ is compensated. Examinations while the patient is asymptomatic may be normal or show only an extrarenal pelvis; the use of a diuretic renogram or a urographic study during acute symptoms has been advocated to demonstrate the underlying abnormality.
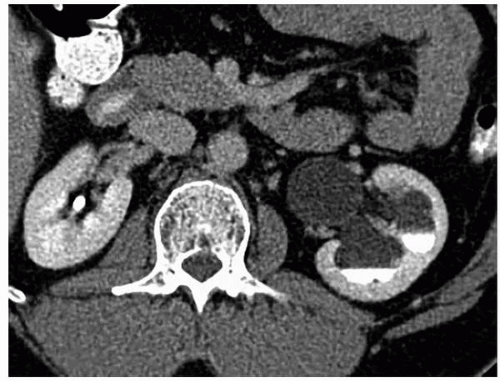 FIGURE 1.13. UPJ obstruction. Enhanced CT in excretory phase demonstrates a dilated left renal pelvis and caliectasis with layering of contrast in the dependent portions of the collecting system. |
The renal pelvis and calyces are dilated, and with long-standing or very high-grade obstruction, a virtually nonfunctioning kidney may be present. The UPJ itself, which may be difficult to visualize, appears to join the renal pelvis in a higher or more medial position (Figs. 1.13 and 1.14). Care must be taken to differentiate a true UPJ obstruction (Fig. 1.15) from a large extrarenal pelvis. In the
latter case, the renal pelvis may appear quite dilated; however, in the absence of caliectasis, the diagnosis of UPJ obstruction should not be entertained. Variable filling of the ureter may occur distal to the UPJ. When there is significant kinking of the UPJ, an aberrant vessel may be the cause of the obstruction.
latter case, the renal pelvis may appear quite dilated; however, in the absence of caliectasis, the diagnosis of UPJ obstruction should not be entertained. Variable filling of the ureter may occur distal to the UPJ. When there is significant kinking of the UPJ, an aberrant vessel may be the cause of the obstruction.
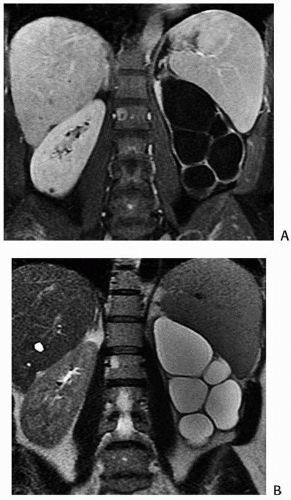 FIGURE 1.14. UPJ obstruction. MR images in T1 (A) and T2 weighting (B) demonstrate markedly dilated calyces. |
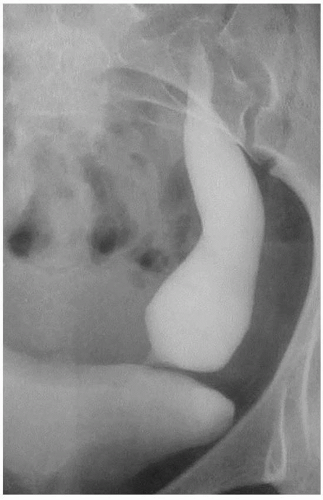 FIGURE 1.15. Primary megaureter. Excretory urogram demonstrates a markedly dilated distal left ureter. |
Pyeloplasty has been considered the treatment of choice for UPJ obstruction. Percutaneous nephrostomy followed by balloon pyeloplasty or by endoscopic endopyelotomy has been reported as a primary procedure and as a secondary procedure in patients in whom surgical therapy has failed. Although success rates of up to 85% have been reported with endopyelotomy, pyeloplasty consistently corrects the defect in 85% to 90% of patients. Endopyelotomy probably should not be used in infants and has a higher failure rate in patients with extremely redundant renal pelves or long UPJ strictures.
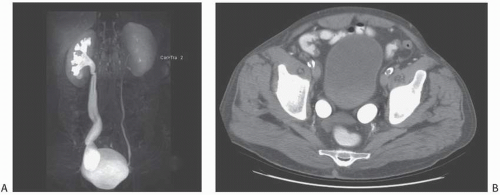 FIGURE 1.16. Primary megaureter. A: MR urogram demonstrates dilated right collecting system and ureter with a “beak” in the distal ureter characteristic of a primary megaureter. (Courtesy of Seog Wan Ko, MD, Department of Radiology, Gwangju Christian Hospital, Gwangju, South Korea.) B: Axial CT image from a different patient showing bilaterally dilated distal ureters. The more proximal ureters were normal indicating primary megaureter as the cause. |
Congenital megaureter, or primary megaureter, is a functional obstruction of the distal ureter. Typically, approximately 2 cm of the ureter just above the ureteral orifice is involved, and this abnormal segment is usually of normal caliber. However, the ureter proximal to this improperly functioning segment is variably dilated. It is not unusual to see this dilation involving only the distal one-third of the ureter, although if the obstruction is severe enough, significant hydronephrosis may occur (Fig. 1.16). As with UPJ obstruction, a normal-size ureteral catheter can be easily passed through the abnormal ureteral segment. Thus, this narrowing is not an anatomic stricture; rather, it is a failure of the abnormal ureteral segment to undergo normal peristalsis. It is also possible that the abnormality is a very mild narrowing or an inability to distend to the maximal normal diameter of the most distal part of the ureter. Under these conditions, every bolus, which is pushed down the ureter by a following contraction wave, has its hydrostatic pressure rise above normal as it is pushed through the limited-distensibility terminal segment. Over time, the ureter just above this segment dilates and, as any other significantly dilated ureter, is no longer capable of transmitting a normal contraction wave. Treatment for severe cases includes excision of the abnormal distal segment, surgical tapering of the dilated “normal” ureter, and reimplantation of the tapered ureter into the bladder using antirefluxing techniques.
Circumcaval Ureter
The embryology of the IVC is complex. Normally, the suprarenal IVC derives from the right subcardinal vein and the infrarenal IVC derives from the right supracardinal vein. Circumcaval ureter results when the subcardinal vein, which lies ventral to the ureter, persists and forms the major portion of the vena cava (Fig. 1.17). The right ureter is carried medially by the migration of the subcardinal vein toward the developing IVC. Symptoms of circumcaval ureter, if any, relate to the degree of ureteral obstruction.
The typical pattern is a tortuous, dilated proximal right ureter and associated hydronephrosis. The course of the proximal ureter is described as having a “reverse J” configuration before it crosses behind and around the IVC and then descends medial to the ipsilateral lumbar pedicle This severe medial deviation is the hallmark of a circumcaval ureter.
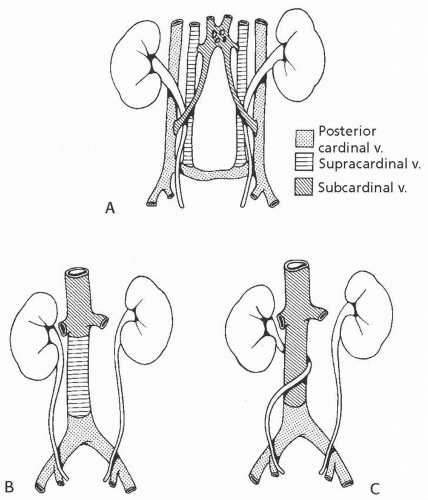 FIGURE 1.17. Embryology of circumcaval ureter. A: Embryologic condition, with ureter winding among three cardinal veins. B: Normal development of vena cava, with infrarenal portion arising from the supracardinal vein that lies under ureter. C: Circumcaval ureter develops when the infrarenal portion of the vena cava develops from the subcardinal vein, which in embryonic life is ventral to the ureter. (Modified from Fig. 9.195 in Witten DM, Myers GH, Utz DC, eds. Emmett’s Clinical Urography. 4th ed. Philadelphia, PA: WB Saunders Co.; 1977:678, with permission.) |
Duplex Collecting Systems
Duplication of the collecting system may be partial or complete. Partial duplication is the most frequently occurring congenital anomaly in the urinary tract. The duplex collecting system and its related abnormalities can result in some of the most complicated patterns detected on imaging studies in both children and adults.
The autopsy incidence of partial duplication is approximately 1 in 150 cases, whereas complete duplication is found approximately once in every 500 cases. However, the incidence in clinical series for this anomaly is up to six times higher, probably reflecting the likelihood for this anomaly to produce symptoms. Complete duplications are bilateral in up to 20% of cases.
Ureteral Anatomy
The UPJ is usually a gentle tapering of the renal pelvis as it joins the proximal ureter. The ureter remains retroperitoneal throughout its course as it crosses anterior to the common or external iliac vessels. It curves laterally and then medially before coursing submucosally in the bladder wall for about 2 cm before entering the bladder lumen at the lateral margin of the trigone. The ureterovesical junction is the narrowest portion of the ureter.
Embryology
The ureter forms as a bud from the mesonephric duct (Fig. 1.18). The normal kidney is formed when this bud invaginates the metanephric blastema and, through multiple branchings, forms the collecting system and distal collecting ducts. Partial duplication results from the branching of the ureteral bud before it connects with the metanephric blastema. The point at which the ureteral bud branches will determine the level at which the two ureters join; this bifurcation may occur anywhere from the bladder wall to the renal pelvis. The latter will result in a bifid renal pelvis. On rare occasions, the single ureteral bud may trifurcate or may even branch into four or five segments before meeting the metanephric blastema, resulting in multiplication of the renal pelvis. Another variation occurs when one branch of a partially duplicated system fails to reach the kidney and becomes a blind-ending stump connected to the functional ureter (Fig. 1.19). This branch is sometimes short and is referred to as a ureteral diverticulum. However, because all layers of the ureteral wall are present, this branch is not a true diverticulum and is more properly termed a blind-ending ureteral bud.
Complete duplication of the ureter occurs when two separate buds arise from the mesonephric duct. These buds invaginate the metanephric blastema separately, resulting in the formation of separate upper and lower intrarenal collecting systems. Each of these systems is known as a moiety, and each is drained by a separate ureter. As the mesonephric duct migrates caudally during embryonic life, the ureter from the lower moiety is deposited near its expected normal location in the bladder, and its ureterovesical junction is therefore usually close to its normal position on the trigone. The ureter from the upper moiety remains attached to the mesonephric duct longer during its caudal migration and eventually connects with the bladder inferiorly and medially. The Meyer-Weigert law states that the ureter from the upper moiety will enter the bladder inferiorly and medially (the ectopic ureter) in relation to the ureter draining the lower moiety (the orthotopic ureter). The corollary to the Meyer-Weigert law is that the upper moiety often obstructs while the lower moiety may reflux.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


