The Adrenal Gland
INTRODUCTION
The adrenal glands lie superior to the kidneys in the perinephric space. Since they are surrounded by fat, they are usually well visualized on computed tomography (CT) (Fig. 3.1) or MR examinations. Patients with abnormalities involving the adrenal glands may present with symptoms due to the adrenal disease, or the abnormality may be identified as an incidental finding on an examination performed for another indication. Because the adrenal gland is an active site of synthesis of a variety of hormones, patients may present with symptoms of hormone excess or, less likely, hormone deficiency. Thus, adrenal diseases are discussed as hyperfunctional disorders in which excess hormone is produced, as disorders of adrenal insufficiency, and as diseases in which adrenal function is normal. Because these hormones can be readily measured, there is seldom doubt as to which category a patient belongs.
 FIGURE 3.1. Normal adrenal glands. Both glands have an inverted “Y” configuration, with the tail pointed anteromedially. |
HYPERFUNCTIONAL DISEASES
Cushing Syndrome
Cushing syndrome is the clinical manifestation of excess glucocorticoids. These steroids may come from an exogenous or endogenous source. Exogenous Cushing syndrome is seen in patients treated with large doses of steroids. Endogenous Cushing syndrome is caused by overproduction of cortisol by the adrenal cortex. The characteristic appearance of a patient with Cushing syndrome includes truncal obesity, hirsutism, abdominal striae, and muscle atrophy. Hypertension and glucose intolerance are common findings. The diagnosis of Cushing syndrome may be confirmed by measuring urinary-free cortisol levels, which are elevated.
Patients may be further classified as having adrenocorticotropic hormone (ACTH)-dependent or ACTH-independent Cushing syndrome. Plasma ACTH levels are elevated in ACTH-dependent and depressed in those with ACTH-independent Cushing syndrome. In approximately 80% of cases, unregulated amounts of ACTH stimulate the adrenal cortex, which results in bilateral adrenal hyperplasia. This is referred to as ACTH-dependent Cushing syndrome. ACTH-independent causes of Cushing syndrome include adrenal adenomas, carcinomas, and two rare hyperplasia syndromes, primary pigmented nodular adrenal dysplasia (PPNAD) and ACTH-independent macronodular hyperplasia (AIMAH).
Among patients with ACTH-dependent Cushing syndrome, the pituitary gland is not always the source of ACTH. A variety of other tumors, such as oat cell carcinoma, bronchial adenoma, and tumors of the ovary, pancreas, thymus, and thyroid, may rarely secrete ACTH.
The etiology of ACTH-independent Cushing syndrome includes an adrenal adenoma, adrenal carcinoma, and several inherited syndromes including multiple endocrine neoplasia type 1 (MEN1), McCune-Albright syndrome, familial adenomatous polyposis, and Carney complex. As many as 40% of patients with MEN1 may have adrenal lesions, usually bilateral nodular hyperplasia. The McCune-Albright syndrome, which usually presents in the first year of life, includes bony lesions, skin pigmentation, precocious puberty, and bilateral nodular hyperplasia. Patients with familial adenomatous polyposis have extensive colonic polyps as well as desmoid tumors, osteomas, retinal abnormalities, and adrenocortical lesions including adenomas, carcinomas, and hyperplasia. Carney complex includes myxomas of the heart, breast, and skin as well as psammomatous melanotic schwannomas and osteochondromyxomas. PPNAD is the most common adrenal lesion reported in these patients. A few patients with adrenal hyperplasia have macronodules that can be seen in the adrenal glands on CT examination. These macronodules usually measure <3 cm and may be <1 cm in diameter. Macronodular hyperplasia is caused by an ACTH-secreting pituitary microadenoma in the majority of cases. Macronodular hyperplasia with a dominant nodule may be distinguished from a cortisol-producing adenoma by the size of the contralateral adrenal gland. In patients with an adenoma, the contralateral gland and other limbs of the ipsilateral gland are atrophic, whereas they are enlarged (hyperplastic) in patients with macronodular hyperplasia.
Primary Aldosteronism
Primary aldosteronism, or Conn syndrome, is the result of excess aldosterone produced by the adrenal glands. Approximately 60% of cases of Conn syndrome are due to hyperplasia and 35% are caused by an adenoma. Rare causes include an adrenal cortical carcinoma and unilateral hyperplasia. Adrenal hyperplasia may be further subdivided into idiopathic hyperaldosteronism (IHA) and primary adrenal hyperplasia (PAH). IHA is far more common than PAH. Since bilateral adrenalectomy does not effectively control the hypertension and hypokalemia in patients with IHA, they are treated medically. Although morphologically PAH resembles IHA, PAH may be unilateral or bilateral, and adrenalectomy is an effective treatment for unilateral cases. Rarely does a primary adrenocortical carcinoma secrete enough aldosterone to cause a recognizable clinical syndrome. As diagnostic advances result in more cases of primary aldosteronism being found in hypertensive patients, a greater portion of these patients have IHA, which should be treated medically.
The syndrome Conn described in 1955 includes hypokalemia, hypertension, elevated serum aldosterone, but low serum renin levels. Excess levels of aldosterone cause sodium retention, an increase in the plasma volume, and hypertension. Because potassium is exchanged for sodium in the distal tubule, sodium retention creates hypokalemia. Aldosteronism occurs also in patients with renovascular hypertension. However, this form of secondary aldosteronism is distinguished from primary aldosteronism by measuring the serum renin level, which is low in patients with Conn syndrome.
Laboratory tests can also be used to help distinguish between hyperplasia and adenoma, as the etiology of primary aldosteronism. However, these tests are not entirely accurate, and radiographic confirmation is needed. Adrenal venous sampling (AVS) is the best method to identify a unilateral source of aldosterone secretion. Although technically challenging, in experienced centers, AVS is highly accurate. The combination of anatomic imaging with CT or MRI and a localizing AVS resulted in a cure rate of >95% after unilateral adrenalectomy.
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders that result in the absence or decreased synthesis of cortisol. The defect may lie in any of the five enzymatic steps necessary to convert cholesterol to corticosteroid, but >90% of CAH cases are due to a deficiency of 21-hydroxylase. The loss of normal feedback inhibition causes the pituitary gland to secrete excess ACTH, which overstimulates the adrenal glands and causes bilateral hyperplasia.
Because androgens do not require 21-hydroxylase for their synthesis, they are overproduced in response to high levels of ACTH. The clinical manifestation depends on the sex of the patient and the age at which the androgen excess appears. Ambiguous genitalia and subsequent virilization are seen in women, whereas precocious puberty is found in young boys.
Virilizing Tumors
Androgen-producing tumors are rare, may be benign or malignant, and occur in either males or females at any age. Patients whose virilization is the result of a tumor usually present at an older age than those with CAH.
The typical clinical manifestations include amenorrhea, hirsutism, enlargement of the clitoris, and deepening of the voice. Elevated testosterone levels are also frequently found in adrenal tumors, so a high testosterone level cannot be used to distinguish a gonadal tumor from an adrenal tumor.
Feminizing Tumors
Feminizing tumors are quite rare, and most are malignant. They are usually seen in men but have also been reported in prepubertal girls and postmenopausal women. Gynecomastia is the predominant clinical manifestation.
Adrenal Insufficiency
Adrenal insufficiency may be caused by tissue destruction of the adrenal glands (primary) or may result from inadequate stimulation by ACTH (secondary). Secondary adrenal insufficiency is more common than primary, and both are found more often in women. Because normal adrenal function depends on ACTH, any disorder that impairs the ability of the pituitary gland to secrete ACTH may lead to adrenal hypofunction. With decreased ACTH activity, cortisol and adrenal androgen secretion diminish, but aldosterone secretion remains relatively intact. Thus, patients with hypopituitarism can tolerate sodium deprivation better than patients with primary adrenal insufficiency. Furthermore, hypopituitary patients do not develop mucocutaneous hyperpigmentation, as this depends on excessive ACTH secretion.
Primary adrenal insufficiency, or Addison disease, occurs only after at least 90% of the adrenal cortex has been destroyed. Idiopathic adrenal atrophy, an autoimmune disorder, is the most common cause of Addison disease in the United States. The other common cause of Addison disease is destruction of the adrenal glands by a granulomatous disease, usually tuberculosis. Among the other causes of adrenal insufficiency, hemorrhage and destruction of adrenal tissue by metastatic tumor are reported most commonly.
The clinical onset of Addison disease usually is gradual and may be difficult to recognize. Manifestations are due to the deficiency of cortisol and aldosterone. Because cortisol deficiency results in increased pituitary secretion of ACTH and other melanocyte-stimulating hormones, patients with Addison disease develop a characteristic hyperpigmentation. Treatment includes cortisol and aldosterone replacement as well as saline to correct extracellular fluid depletion.
The radiographic manifestations of primary adrenal insufficiency depend on the cause of adrenal dysfunction. The plain abdominal radiograph may reveal adrenal calcification commonly seen in tuberculosis or histoplasmosis.
The most useful examinations are CT or MR, which define the size and shape of the adrenal glands. In patients with autoimmune disease, severe cortical atrophy is present, such that the adrenal glands may be difficult to detect.
Granulomatous involvement by tuberculosis or histoplasmosis is bilateral. The glands are enlarged but often maintain a normal configuration. Calcification is common.
If adrenal hemorrhage is acute, it may be recognized by the increased density of the recent hemorrhage. As the density of the hematoma decreases, it becomes indistinguishable from other adrenal masses. Although adrenal metastases are common, adrenal insufficiency infrequently occurs. This is because so much of the adrenal cortex must be destroyed before insufficiency ensues.
Radiology
Ultrasound
Although the normal adrenal gland may be difficult to image with ultrasound, tumors >2 cm often can be detected. The right adrenal gland usually is easier to study because the liver provides a good acoustic window. Ultrasound may be valuable in distinguishing a solid mass from an adrenal cyst.
Computed Tomography
The modality routinely used to evaluate the adrenal glands is CT. The perinephric fat present in most patients allows the adrenal glands to be clearly defined, and tumors as small as 5 mm can be detected. Intravenous contrast medium is seldom needed but may be helpful to distinguish an adrenal mass from the upper pole of the kidney.
The adenomas in Conn syndrome usually are the most difficult adrenal tumors to detect because they tend to be the smallest, averaging <2 cm in diameter (Fig. 3.2).
Patients with Cushing syndrome, conversely, are relatively easy to examine by CT. The tumors in these patients are larger than those causing Conn syndrome, and the adrenal glands are depicted clearly by the abundant retroperitoneal fat (Fig. 3.3). Tumors that are >4 cm in diameter or that demonstrate central necrosis are suspicious for malignancy.
Bilateral adrenal hyperplasia is seen as enlargement of the entire adrenal gland, but without a focal mass. The limbs of both adrenal glands are thickened and elongated (Figs. 3.4 and 3.5). Patients whose hyperplasia is caused by an ectopic source of ACTH tend to have larger adrenal glands than those with a pituitary adenoma as the source of ACTH.
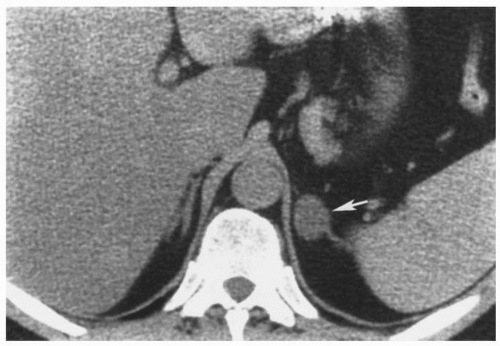 FIGURE 3.2. Adrenal adenoma. An aldosterone-secreting adenoma (arrow) is seen in the left adrenal gland. |
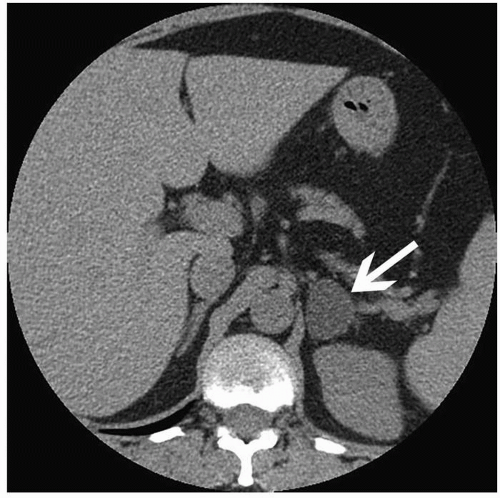 FIGURE 3.3. Adrenal adenoma. A cortisol-secreting adenoma (arrow) is seen in a patient with Cushing disease. |
Enlargement of the adrenal gland usually is more obvious in patients with Cushing syndrome than in those with Conn syndrome. However, there is significant overlap with the appearance of normal adrenal glands, and hyperplasia should not be diagnosed by morphology alone.
A minority of patients with Cushing disease have macronodular hyperplasia in which one or more nodules may be seen (Fig. 3.6). Rarely, patients may have AIMAH in which the large size of the adrenal nodules suggests bilateral adenomas or metastases. PPNAD is another rare cause of ACTH-independent Cushing syndrome. PPNAD is transmitted as an autosomal dominant trait and is associated with the Carney complex. Histologically, pigmented nodules,
2 to 5 mm in diameter, are seen. Patients with AIMAH and PPNAD are treated with bilateral adrenalectomy.
2 to 5 mm in diameter, are seen. Patients with AIMAH and PPNAD are treated with bilateral adrenalectomy.
 FIGURE 3.4. Bilateral adrenal hyperplasia. Both adrenal glands are elongated and thickened. |
 FIGURE 3.5. Bilateral adrenal hyperplasia. Both adrenal glands are seen as thickened on this coronal reconstructed CT image. |
Venous Sampling
Because each adrenal gland is drained by a central vein, the venous efflux of each gland can be sampled for hormone analysis. Adrenal venous sampling (ADV) can be valuable in patients with Conn syndrome because the aldosterone-secreting adenoma often is <1 cm and may be too small to be detected by CT. Furthermore, patients may have PAH or a nonhyperfunctioning adenoma, which cannot be distinguished from an adenoma secreting excess aldosterone. Localizing the side of excess hormone production by venous sampling guides surgical adrenalectomy. Venous sampling is needed rarely in evaluating patients with Cushing syndrome but may be applied to those patients suffering from masculinizing or feminizing tumors.
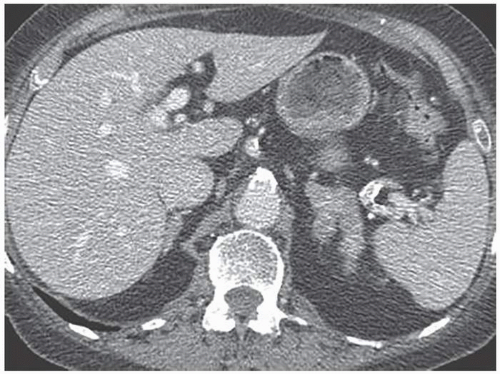 FIGURE 3.6. Bilateral macronodular hyperplasia. Multiple small nodules are seen in the left adrenal gland. Only the top of the right adrenal gland is shown, but it also contains multiple nodules. |
AVS is technically difficult due to the small size of the adrenal veins. Since the left adrenal vein empties into the left inferior phrenic vein, it is easier to find and sample. The right adrenal vein empties directly into the inferior vena cava posteriorly in most patients but may have a common trunk with a hepatic vein in a significant minority of cases. Finely collimated CT scanning in the venous phase of contrast enhancement allows visualization of the right adrenal vein in most patients.
Magnetic Resonance
The perinephric fat provides excellent contrast for magnetic resonance imaging (MRI) as well as for CT. The spatial resolution of MRI is similar to that of CT, and the additional parameters available to distinguish benign from malignant lesions may be useful. For most patients with hyperfunction of the adrenal cortex, MRI does not provide additional information after a CT examination.
ADRENAL MEDULLA
Pheochromocytoma
Pheochromocytomas are tumors composed of chromaffin cells and usually are located in the adrenal medulla. They are rare tumors and account for <1% of patients with systemic hypertension. Extraadrenal pheochromocytomas are called paragangliomas and may be divided into those that arise from parasympathetic tissues along the cranial or vagus nerves and those that arise from sympathetic chromaffin tissue. Thus, they may lie anywhere between the base of the brain and the epididymis but usually lie along the sympathetic chain in the retroperitoneum. Eighty-five percent of pheochromocytomas arise from chromaffin tissue in the adrenal medulla. Sporadic pheochromocytomas average 4 cm in diameter at presentation, while those associated with syndromes are often smaller.
Patients may complain of episodes of headaches associated with palpitation and diaphoresis. If the pheochromocytoma is located in the bladder wall, micturition may induce a symptomatic episode.
Hypertension is the most common finding and is present in >90% of patients. Hypertension from a pheochromocytoma may be difficult to distinguish from renovascular or essential hypertension; however, patients with a pheochromocytoma are more likely to have labile hypertension with discrete paroxysmal attacks. These patients are also prone to hypertensive attacks during anesthesia induction. Manipulation of the gland during surgery, percutaneous adrenal biopsy, or even selective adrenal angiography may also induce a hypertensive crisis.
Pheochromocytomas are associated with other endocrine tumors. The multiple endocrine neoplasia syndrome 2A (MEN2A) includes medullary carcinoma of the thyroid and parathyroid hyperplasia as well as pheochromocytoma. The MEN2B syndrome is rare and is composed of pheochromocytoma; medullary carcinoma of the thyroid; and the mucocutaneous manifestations of mucosal neuromas, intestinal ganglioneuromatosis, and a marfanoid habitus. The majority of patients with the MEN2A or the MEN2B syndrome have pheochromocytomas that are bilateral and usually intra-adrenal. However, all manifestations of the syndrome may not occur at the same time. Thus, a careful history of a previous endocrine abnormality should be obtained in evaluating patients who may fall into these categories. These syndromes are inherited in an autosomal dominant fashion.
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2A
Medullary carcinoma of thyroid
Pheochromocytoma (usually bilateral)
Parathyroid adenoma
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2B
Medullary carcinoma of thyroid
Pheochromocytoma (usually bilateral)
Mucosal neuromas
Intestinal ganglioneuromatoses
Marfanoid habitus
Pheochromocytoma is also associated with neurofibromatosis (type I) and von Hippel-Lindau syndrome (type II). Paragangliomas are also associated with tuberous sclerosis and Sturge-Weber syndrome.
A syndrome of familial pheochromocytomas not associated with other endocrine tumors has also been noted. Specific gene mutations have been identified involving the mitochondrial enzyme succinate dehydrogenase (SDH). The most common of these are SDHB mutations, where patients develop thoracic or abdominal paragangliomas with a propensity for malignancy. Patients with SDHD mutations develop paragangliomas in the head and neck.
In 1977, Carney et al. reported the association of a gastric leiomyosarcoma, a pulmonary chondroma, and a functioning extraadrenal pheochromocytoma. This rare combination of neoplasms often is termed Carney triad.
Although 85% of all pheochromocytomas are located in the adrenal medulla, there is a striking difference between sporadic tumors and those associated with the MEN syndromes. The sporadic pheochromocytomas are located outside the adrenal gland in as many as 25% of cases, whereas those associated with the MEN2 syndromes are almost always intra-adrenal. Pheochromocytomas found in patients with MEN2 syndrome are usually multicentric and involve both adrenal glands in >80% of cases.
The histologic appearance also differs in sporadic and MEN2-associated cases. In sporadic cases, the tumor is well encased with normal adjacent medulla. In patients with MEN2 syndrome, the medulla is hyperplastic and the tumor may be multicentric.
If a pheochromocytoma is suspected, the diagnosis can be made by measuring elevated levels of serum or urine catecholamines. Urinary metanephrine or vanillylmandelic acid (VMA) is elevated in >90% of patients when measured on 24-hour urine collections. Plasma-free metanephrine levels are elevated in 99% of patients. However, several separate determinations should be made because of the episodic hormone secretion found in these patients. Furthermore, patients taking medications such as methyldopa or methenamine may have falsely high catecholamine levels.
Approximately 15% of pheochromocytomas lie outside the adrenal gland. Those which arise from sympathetic paraganglia are called paragangliomas, whereas those which arise from the sympathetic tissue of the cranial or vagus nerves are glomus tumors, chemodectomas, or carotid body tumors. Although they may occur anywhere from the base of the skull to the epididymis, the most common locations are in the sympathetic chain in the retroperitoneum from the level of the adrenal glands to the aortic bifurcation, which includes the region of the organ of Zuckerkandl, which lies near the origin of the inferior mesenteric artery.
Pheochromocytomas usually are benign, but approximately 13% demonstrate malignant behavior. Extra-adrenal pheochromocytomas are more likely to be malignant than intra-adrenal tumors.
A small number of patients have a pheochromocytoma that does not produce sufficient catecholamine to cause clinical symptoms. These “nonfunctioning” pheochromocytomas are discovered as palpable masses or as incidental findings at surgery and at autopsy or on imaging studies such as CT or MR examinations. In a recent series by Motte-Ramirez et al., more than half of their series of pheochromocytomas were discovered incidentally. The nonfunctioning tumors tend to be larger than functioning pheochromocytomas whose hormone production brought them to attention earlier than patients with nonfunctioning pheochromocytomas.
The treatment for patients with pheochromocytoma is surgical resection. Biochemical confirmation of the diagnosis and accurate preoperative radiologic localization have made these operations much safer. Nevertheless, patients undergoing surgical resection still must receive adrenergic blockade before the induction of anesthesia as well as careful monitoring during the operation to treat any crises. Both an α-adrenergic blocker (phenoxybenzamine) as well as a β-blocker (propranolol) are advocated, whereas nitroprusside may be used to treat hypertensive episodes. This same regimen may be used in patients requiring invasive radiologic procedures.
Radiology
Ultrasound
Ultrasound may be used to localize an intra-adrenal pheochromocytoma, but also it may identify ectopic tumors lying in the paraaortic area. Pheochromocytomas tend to be more echogenic than normal adrenal tissue, possibly because of the hypervascularity they usually exhibit. Ultrasound is also often helpful in children, in whom the relative lack of retroperitoneal fat makes CT evaluation difficult.
Computed Tomography
CT is the primary imaging modality used to localize a pheochromocytoma (Figs. 3.7 and 3.8). Most intra-adrenal pheochromocytomas are detected, and a sensitivity of 95% can be expected. Care must be taken when examining patients with the MEN syndrome, however, because the tumors in these patients often are small (Fig. 3.9). Because most extra-adrenal pheochromocytomas (paragangliomas) lie in the para-aortic region, they are also demonstrated on an abdominal CT examination (Fig. 3.10).
The administration of intravascular contrast media is unnecessary for most patients. The adrenal glands are well visualized, and most tumors are large enough to be readily identified, measuring 2 to 5 cm in diameter at presentation. Areas of necrosis or
hemorrhage are common, especially in larger tumors. Although the intravenous administration of ionic contrast media in patients with a pheochromocytoma has been shown to stimulate sufficient catecholamine secretion to cause a hypertensive crisis, this does not appear to be the case with nonionic contrast media.
hemorrhage are common, especially in larger tumors. Although the intravenous administration of ionic contrast media in patients with a pheochromocytoma has been shown to stimulate sufficient catecholamine secretion to cause a hypertensive crisis, this does not appear to be the case with nonionic contrast media.
 FIGURE 3.7. Pheochromocytoma. A 3-cm left adrenal mass just anterior to the kidney shows heterogeneous contrast enhancement. |
 FIGURE 3.8. Pheochromocytoma. A 4-cm right adrenal pheochromocytoma demonstrates heterogeneous enhancement on this T1-weighted MR examination. An incidental hepatic hemangioma is present. |
 FIGURE 3.9. MEN syndrome. This patient with a history of medullary carcinoma of the thyroid had a right pheochromocytoma removed (surgical clips) 2 years earlier. A small left adrenal mass (arrow) is detected on this unenhanced CT scan. |
 FIGURE 3.10. Paraganglioma. A 2-cm mass (arrow) is seen on this enhanced abdominal CT examination. |
Magnetic Resonance Imaging
MRI has a high sensitivity for the detection of pheochromocytomas. The tumor is typically hypointense to the liver on T1-weighted images and may have a very high signal intensity on T2-weighted images. However, the most common appearance on T2-weighted images is an enhancing, heterogeneous mass with focal areas of high signal intensity (Fig. 3.11).
Nuclear Medicine
Radionuclide examinations using iodine-131-labeled metaiodobenzylguanidine (MIBG) or indium-111-labeled pentetreotide (Octreoscan) may be very useful in localizing pheochromocytomas or paragangliomas (Fig. 3.12).
The overall accuracy of MIBG is similar to that of CT and MRI; however, scintigraphy has several advantages. With a single injection of radionuclide, the entire body can be scanned. This is particularly helpful for ectopic tumors or for the detection of metastatic deposits. MIBG can also detect medullary hyperplasia, which is seen in patients with MEN syndromes and may be an early manifestation of a developing pheochromocytoma.
Positron emission tomography (PET) with 2-[fluorine-18] fluoro-2-deoxy-D-glucose (FDG) may also be used to localize pheochromocytomas. FDG is accumulated by the majority of both benign and malignant pheochromocytomas, including some that fail to concentrate MIBG.
Neuroblastoma and Ganglioneuroma
Neuroblastomas are primitive tumors that arise from sympathetic nervous system tissue. They may occur in the neck, thorax, abdomen, and pelvis, but no definite primary site can be found in a significant minority of patients. The most common location is the adrenal gland, which accounts for approximately 35% of patients.
Neuroblastomas usually occur in young children. Approximately 25% of cases occur in the first year of life, and as many as 60% occur by the age of 2 years.
Although neuroblastoma is the most common extracranial malignant tumor in childhood, its incidence is only 1 to 3 per
100,000 children per year. An increased incidence is seen in neurofibromatosis, and familial associations have been reported.
100,000 children per year. An increased incidence is seen in neurofibromatosis, and familial associations have been reported.
 FIGURE 3.11. Pheochromocytoma. The right adrenal mass shows heterogeneous enhancement on this MR examination. |
 FIGURE 3.12. Pheochromocytoma. A: The posterior view of this 123-iodine MIBG scan demonstrates intense MIGB uptake in the right posterior abdomen and physiologic activity in the liver. SPECT (B) and fused SPECT/CT (C) scans demonstrate an MIBG-avid right adrenal nodule consistent with pheochromocytoma. |
Some neuroblastomas spontaneously mature into benign ganglioneuromas. Histologically, neuroblastomas contain densely packed small round cells that may be difficult to differentiate from other tumors, such as Ewing sarcoma, lymphoma, or rhabdomyosarcoma. Tumors that contain more mature ganglion cells mixed with neuroblasts are classified as ganglioneuroblastomas. Ganglioneuroblastomas and ganglioneuromas are differentiated from neuroblastomas by a greater degree of cellular maturation. Ganglioneuroblastomas are malignant tumors but may be partially or totally encapsulated. Ganglioneuromas are benign tumors with an intact capsule, but careful evaluation of the entire tumor is necessary because there may be marked variation in different parts of the tumor.
Ganglioneuromas are benign, nonhyperfunctioning tumors composed of mature ganglion cells and Schwann cells in a fibrous stroma. They arise from the sympathetic nervous system and may be found in the neck, thorax, abdomen, or pelvis. In one series, 19 (41%) of 46 abdominal ganglioneuromas were found in the adrenal gland. Because they are not hormonally active, they are usually found as a large mass or as an incidental finding on CT or MRI examinations.
An adrenal ganglioma is seen as a well-defined, rounded, homogeneous mass with an attenuation slightly less than muscle on unenhanced CT images. With the administration of intravascular contrast media, mild heterogeneity may be seen, especially in larger tumors, but the attenuation still usually is less than muscle (Fig. 3.13).
A homogeneous tumor of relatively low signal intensity is seen on T1-weighted MR images. A heterogeneous high signal intensity has been reported on T2-weighted images.
The most common presentations of a child with a neuroblastoma are pain, abdominal distention, or an abdominal mass discovered by a parent. Most patients have elevated urinary catecholamines when measured as catechol excretion per milligram of creatinine. More than 50% of patients excrete high levels of VMA, and up to 90% of patients have an elevation of VMA or homovanillic acid.
 FIGURE 3.13. Ganglioneuroma. This retroperitoneal mass (G) was detected as an incidental finding on an enhanced CT examination. |
The International Neuroblastoma Staging System (Table 3.1) is based on clinical, radiologic, and surgical features. Stage I tumors are those that have no nonadherent positive lymph nodes or metastases and can be completely resected. Tumors that invade one side of the neural canal are stage II. Tumors that cross the midline to the opposite side of the vertebral body are stage III. Stage IV tumors are those with distant metastases, whereas stage IVs tumors are those in which the tumor cells represent fewer than 10% of all marrow cells; other metastases are limited to the liver and skin. Approximately 70% of patients have metastases at the time of presentation.
Radiology
Ultrasound
Ultrasound is valuable especially in small children who have a paucity of retroperitoneal fat. A mass of heterogeneous echogenicity usually is seen in the region of the adrenal gland (Fig. 3.14). The
margins are poorly defined, and a capsule is not present. A sonographic “lobule” of homogeneous increased echogenicity consisting of an aggregate of cells separated from the surrounding tumor by collagen deposition has been described in a minority of patients. Ultrasound can also be used to detect involvement of adjacent vascular structures, such as the inferior vena cava.
margins are poorly defined, and a capsule is not present. A sonographic “lobule” of homogeneous increased echogenicity consisting of an aggregate of cells separated from the surrounding tumor by collagen deposition has been described in a minority of patients. Ultrasound can also be used to detect involvement of adjacent vascular structures, such as the inferior vena cava.
TABLE 3.1 Neuroblastoma Staging | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 FIGURE 3.14. Neuroblastoma. A: Axial Doppler ultrasound demonstrates a retroperitoneal mass (arrow) displacing the inferior vena cava. B: Enhanced CT images demonstrate an ill-defined lobular mass (white arrows) encasing the inferior vena cava (short black arrow) and the aorta (long black arrow). |
 FIGURE 3.15. Neuroblastoma. Calcification is seen in this malignant tumor involving para-aortic lymph nodes. |
 FIGURE 3.16. Neuroblastoma. A: Para-aortic masses are present but are difficult to distinguish on an unenhanced CT examination. B: After contrast injection, the kidneys and major retroperitoneal vessels are clearly defined. |
Computed Tomography
Unenhanced CT scans are more sensitive than plain radiographs in detecting the mottled calcification commonly seen in neuroblastoma (Fig. 3.15). Intravenous contrast medium commonly is used to distinguish a renal from an extrarenal mass (Fig. 3.16). CT may be used to identify neuroblastomas in the abdomen, pelvis, or chest. Involvement of adjacent organs or retroperitoneal lymph nodes can be detected.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


