7 Congenital Brain Diseases
Phacomatoses
Neurofibromatosis (Figs. 7.1, 7.2)
Neurofibromatosis Type 1
Frequency: a common phacomatosis (with a prevalence of one in 3000 in Central Europe).
Suggestive morphologic findings: café-au-lait pigmentation, sphenoid wing dysplasia, gliomas (especially optic glioma), and neurofibromas.
Procedure: postcontrast examination is essential.
Other studies: magnetic resonance imaging (MRI) is the imaging modality of choice.
Checklist for scan interpretation:
 Multiple meningiomas?
Multiple meningiomas?
 Acoustic schwannoma?
Acoustic schwannoma?
 Glioma?
Glioma?
 Pathogenesis
Pathogenesis
Neurofibromatosis is a phacomatosis (neurocutaneous syndrome) with an autosomaldominant mode of inheritance and approximately 100% penetrance. The mutated gene is located on chromosome 17, and the spontaneous mutation rate is high (approximately 50% of new somatic mutations).
 Frequency
Frequency
Neurofibromatosis type 1, formerly known as von Recklinghausen disease, is the most common phacomatosis.
 Clinical Manifestations
Clinical Manifestations
A distinction is drawn between overt and abortive forms of the disease. The overt forms can be diagnosed from the presence of café-au-lait spots or neurofibromas. Optic nerve glioma is often the initial manifestation of the less overt forms.
 CT Morphology
CT Morphology
Computed tomography (CT) often demonstrates marked sphenoid wing dysplasia, which tends to be unilateral and can extend to complete absence of the bone. Optic glioma appears as a thickening of the optic nerve that extends into the optic chiasm. Intraorbital neurofibromas are common and can assume various forms. A series of masses several millimeters in diameter arranged in a string-of-beads pattern is a very characteristic finding. The frequent extracoronal location is clearly demonstrated by MRI and by special CT views.
 Differential Diagnosis
Differential Diagnosis
Differentiation from neurofibromatosis type 2 is straightforward, owing to the distinctive character of the tumors. Craniosynostosis is another common setting in which sphenoid wing dysplasia may occur. Optic neuritis is often considered initially in patients with an isolated optic nerve glioma, but persistence or rapid enlargement of the lesion should raise suspicion of a glioma. Involvement of the optic chiasm further supports the diagnosis of optic glioma rather than neuritis. Sarcoidosis is a potential source of confusion, but is very rare. With intraorbital neurofibromas, it is often necessary to consider numerous diagnostic possibilities. Multiple tumors in the same patient can show varying enhancement characteristics after contrast administration. Lesions at certain sites can even mimic cavernomas.
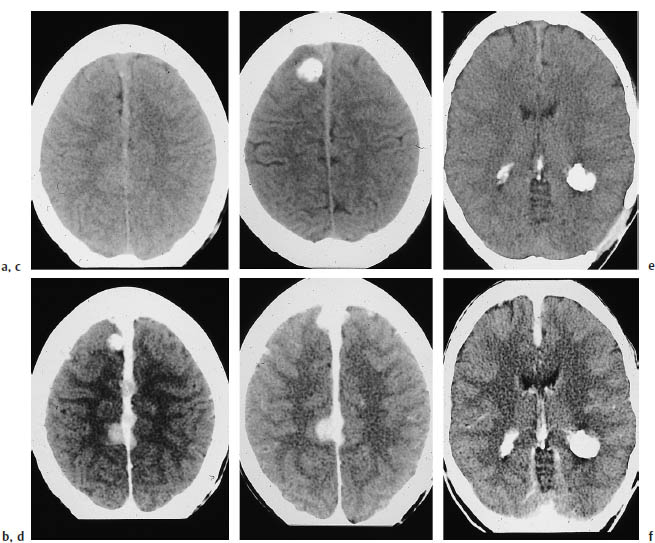
a, c Noncontrast CT shows calcification of the lesions.
b, d Postcontrast CT shows multiple enhancing tumors on the thickened falx.
e, f Large calcified tumors are visible in the occipital horns of both lateral ventricles. Even here, meningiomas and physiologic plexus calcification cannot be excluded as the cause.
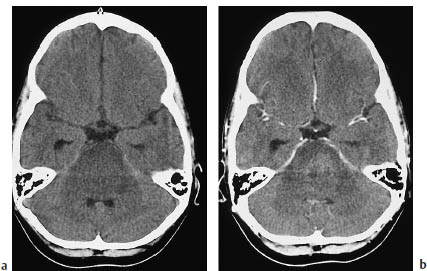
a Before contrast administration.
b fter contrast administration.
Neurofibromatosis Type 2
Frequency: less common than neurofibromatosis type 1.
Suggestive morphologic findings: bilateral acoustic schwannomas; may coexist with meningioma.
Procedure: obtain thin-slice bone-window CT scans to define the internal auditory canal.
Other studies: typical acoustic schwannomas are visible on CT only when a large extrameatal component is present.
Checklist for scan interpretation:
 Expansion of the internal auditory canal?
Expansion of the internal auditory canal?
 Intrameatal contrast enhancement?
Intrameatal contrast enhancement?
 Pathogenesis
Pathogenesis
Neurofibromatosis type 2 has been recognized as a separate phacomatosis. The tumors in this disease arise from Schwann cells and from the meninges. Cutaneous neurofibromas are not observed. Neurofibromatosis type 2 results from a mutation on chromosome 22.
 Frequency
Frequency
The disease is less common than neurofibromatosis type 1, with an estimated incidence of approximately one in 50,000 births.
 Clinical Manifestations
Clinical Manifestations
The characteristic acoustic schwannomas and meningiomas may be manifested at any age, but the third decade is typical. The symptoms are the same as those associated with acoustic schwannomas and meningiomas that occur outside the setting of neurofibromatosis.
 CT Morphology
CT Morphology
With acoustic schwannoma, expansion of the internal auditory canal on bone-window CT may signify an intrameatal acoustic schwannoma. Since the detection of this enlargement is often based on a side-to-side comparison, the bilateral acoustic schwannomas in neurofibromatosis type 2 can be misleading. Enlargement to more than 8 mm is very suspicious for an intrameatal tumor, and should be investigated by MRI. Large extrameatal tumors are visible as enhancing masses in the cerebellopontine angle. As expected, meningiomas are hyperdense on noncontrast CT, are broadly based on the calvarium, and enhance intensely after contrast administration.
 Differential Diagnosis
Differential Diagnosis
Bilateral acoustic schwannomas, even when accompanied by meningiomas, establish the diagnosis of neurofibromatosis type 2.
Frequency: rare (the prevalence is approximately one in 40000).
Suggestive morphologic findings: hemangioblastoma combined with retinal hemangioma.
Procedure: surgical removal of the intracranial tumor.
Other studies: angiography and MRI are more sensitive than CT for detecting mural nodules.
Checklist for scan interpretation:
 One or more cystic masses with an enhancing nidus?
One or more cystic masses with an enhancing nidus?
 Pathogenesis
Pathogenesis
The von Hippel-Lindau syndrome is thought to have an autosomal-dominant mode of transmission, with incomplete penetrance. The genetic anomaly has recently been identified, and appears to involve a defective tumorsuppressor gene on chromosome 3.
 Frequency
Frequency
Rare (the prevalence is approximately one in 40,000).
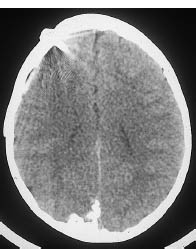
Fig. 7.3 Sturge-Weber syndrome. Sturge-Weber syndrome is characterized by angiomatosis of the cerebral surface. The diagnosis is suggested by corkscrewlike calcifications that follow the cerebral convolutions. This CT scan shows a corkscrew-like calcification on the surface of the right occipital lobe. The disease had been present for some time, prompting the insertion of a ventricular drain.
 Clinical Manifestations
Clinical Manifestations
The two most common tumor entities that are observed in von Hippel-Lindau syndrome differ in their times of appearance: retinal hamartomas occur before puberty, while hemangioblastomas develop after puberty. Hemangioblastomas of the cerebellum lead to gait disturbance or the characteristic features of hydrocephalus. Retinal hamartomas often prompt referral for ophthalmologic examination.
 CT Morphology
CT Morphology
Retinal hamartomas are too small to be detected on CT scans. Hemangioblastomas appear as cystic masses with mural tumor nodules that enhance strongly after contrast administration.
 Differential Diagnosis
Differential Diagnosis
Cystic posterior fossa masses in von Hippel-Lindau syndrome are sometimes difficult to distinguish from pilocytic astrocytomas or cystic metastases from carcinoma.
Sturge-Weber Syndrome (Fig. 7.3)
Frequency: rare.
Suggestive morphologic findings: xtensive vascular malformation projected in the subarachnoid space, appearing either as an enhancing mass or as calcification; tram-track pattern of calcifications.
Procedure: it is occasionally necessary to resect brain areas underlying angiomatous lesions.
Other studies: MRI demonstrates the effects on cerebral tissue.
Checklist for scan interpretation:
 Tram-track calcifications?
Tram-track calcifications?
 Hemiatrophy?
Hemiatrophy?
 Pathogenesis
Pathogenesis
Sturge-Weber syndrome, also known as encephalotrigeminal angiomatosis, is a congenital disease characterized by the development of superficial vascular malformations involving the face and brain.
The histologic features consist of angiomatous changes in the meninges, usually the pia mater. Both arterial and venous malformations can occur. Similar lesions are found in the face. The pathogenesis is not fully established, but presumably involves persistence of primordial vascular structures. Calcifications bordering the vascular malformations develop within the brain substance. Concomitant hemiatrophy of the affected hemisphere is often present.
 Frequency
Frequency
Rare.
 Clinical Manifestations
Clinical Manifestations
The severity of symptoms correlates roughly with the extent of the vascular malformations on the brain surface. Consequently, the CT and MRI findings are useful in making a prognosis. Seizures are an early feature of Sturge-Weber syndrome. With passage of time, focal neurologic symptoms appear. Most patients show some form of developmental delay.
 CT Morphology
CT Morphology
Noncontrast CT may demonstrate gyral calcifications that follow the cortical band. The characteristic vascular malformations are located in the meninges, which follow the sulci. These lesions enhance intensely with intravenous contrast, creating a gyriform pattern of enhancement. The changes may be more or less extensive and may affect one or both hemispheres. Choroidal angiomas have been described, but generally are not detectable by CT. Vascular malformations in extracranial organs are not characteristic.
 Differential Diagnosis
Differential Diagnosis
Facial angioma (port-wine stain) and other extracerebral changes, along with the age of the patient, should minimize problems of differential diagnosis. Posttraumatic calcifications, calcifications secondary to inflammatory disease, etc., may perhaps be considered.
Tuberous Sclerosis (Fig. 7.4)
Frequency: rare.
Suggestive morphologic findings: subependymal nodules, cortical tubers.
Procedure: MRI is required.
Other studies: MRI is more sensitive than CT for detecting cortical tubers.
Checklist for scan interpretation:
 Evidence of tuberous sclerosis?
Evidence of tuberous sclerosis?
 Pathogenesis
Pathogenesis
Tuberous sclerosis is an inherited disease, but its mode of transmission is uncertain. An autosomal-dominant inheritance with low penetrance is assumed.
 Frequency
Frequency
Tuberous sclerosis is a rare phacomatosis. Its prevalence in Central Europe is estimated at one in 50,000.
 Clinical Manifestations
Clinical Manifestations
The following triad characterizes the fully developed clinical picture of tuberous sclerosis:
• Adenoma sebaceum
• Seizures
• Developmental delay
The characteristic adenoma sebaceum consists of a papular facial rash involving the cheeks and the angle between the nose and lower eyelids.
 CT Morphology
CT Morphology
The spectrum of findings in tuberous sclerosis includes retinal hamartomas, which rarely are visible on CT as small calcifications. A more prominent feature is the characteristic subependymal tumors, which may consist of hamartomas or giant-cell astrocytomas. Other findings consist of cortical hamartomas (tubers) and white-matter lesions. Both the tubers and white-matter lesions are hypodense on noncontrast CT, although the tubers may calcify, and in very rare cases may enhance with contrast. The various tumors are differentiated by their location. The tubers are located in the cerebral cortex, and the other lesions occur in the white matter.
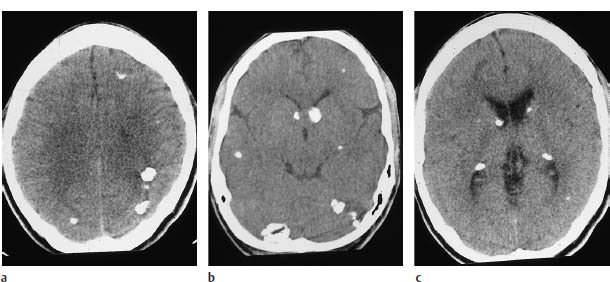
Fig.7.4a–c Tuberous sclerosis.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


