BRONCHIAL ANOMALIES
Anomalies of bronchial anatomy include abnormal origin, absent branches, supernumerary branches, and congenital diverticula (
Table 1-1). Minor variation in subsegmental bronchial anatomy is common but clinically insignificant; a detailed knowledge of subsegmental bronchial anatomy is not necessary for clinical practice. Variation in segmental bronchial anatomy is less frequent and not often of significance.
Tracheal Bronchus
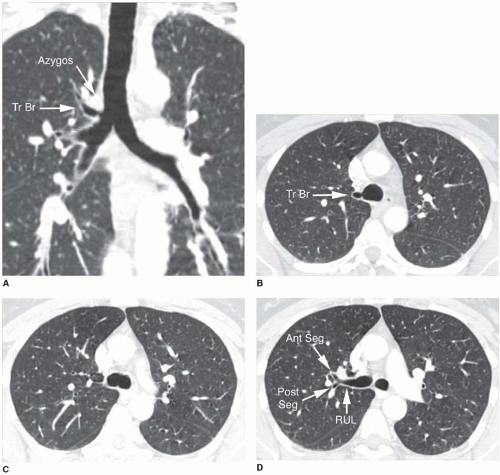
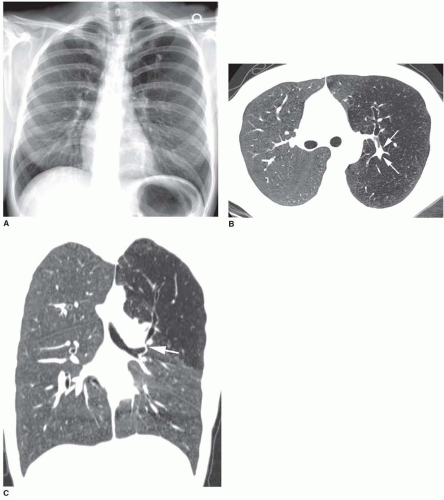
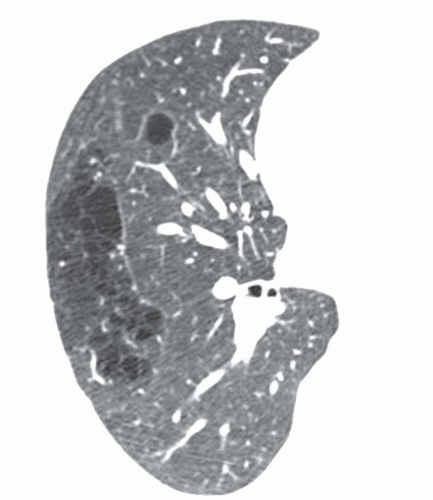
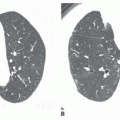
A tracheal bronchus is present in about 0.1% of the population. It usually arises from the right tracheal wall, at or within 2 cm of the tracheal bifurcation. It supplies a variable portion of the medial or apical right upper lobe, most often the apical segment (
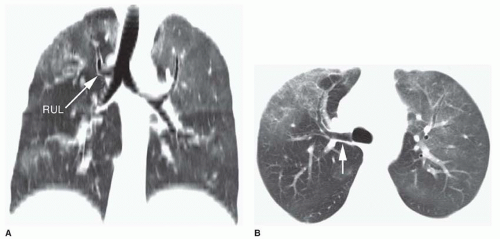
Fig. 1-1); in occasional cases, the entire right upper lobe bronchus arises from the trachea (
Fig. 1-2). When a right-sided tracheal bronchus is present, the azygos arch is seen above the tracheal bronchus. Tracheal bronchus is sometimes referred to as a “pig bronchus” or “bronchus suis” as it is common in pigs and other clovenhoofed animals.
In most cases, this anomaly is insignificant. However, recurrent infection or bronchiectasis may result, since the tracheal bronchus is often slightly narrowed at its origin. A left tracheal bronchus, supplying the apical posterior segment of the left upper lobe, is rarely present; it is much less common than a right-sided tracheal bronchus. The term “tracheal bronchus” is sometimes used to refer to a displaced bronchus, supplying a part of the upper lobe, even if it does not arise from the trachea.
Accessory Cardiac Bronchus
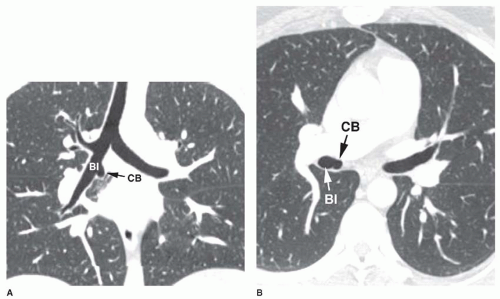
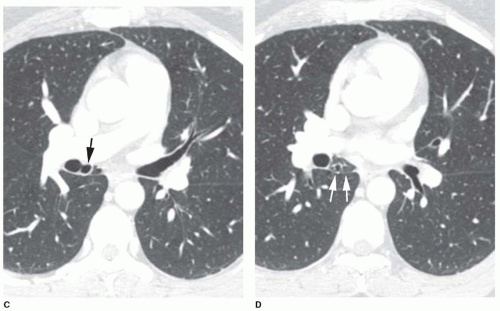
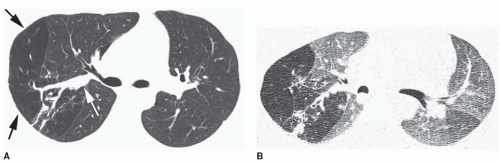
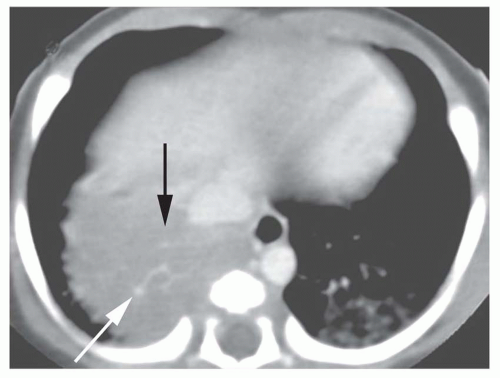
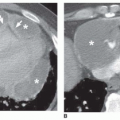
Accessory cardiac bronchus is a supernumerary bronchus with an incidence of about 0.1%. It arises from the medial wall of the bronchus intermedius or right lower lobe bronchus and extends inferiorly and medially toward the mediastinum or heart. In some cases, the cardiac bronchus is a short, blind-ending bronchial stump without associated alveolar tissue, and it may terminate in the mediastinum. In others, a longer branching bronchus is present, associated with rudimentary lung tissue (
Fig. 1-3). In most cases, this anomaly is an incidental finding; occasionally, chronic infection or hemoptysis is associated.
Bronchial Isomerism
Bronchial isomerism refers to bilateral symmetry of the bronchi and associated pulmonary lobes. It may be isolated or associated with a variety of anomalies, particularly congenital heart disease. Bronchial anatomy may be bilaterally right sided (associated with asplenia) or left sided (associated with polysplenia).
BRONCHIAL ATRESIA
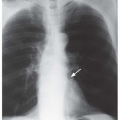
Bronchial atresia is a developmental defect characterized by local narrowing or obliteration of a lobar, segmental, or subsegmental bronchus (
Table 1-2). It is most common in the left upper lobe, followed by the right upper and right middle lobes; it is less common in the lower lobes. This entity is usually detected incidentally in adults and is undoubtedly related to congenital lobar emphysema (CLE). Patients usually have no symptoms, but lung distal to the obstruction may occasionally become infected. In patients with chronic infection, resection may be necessary.
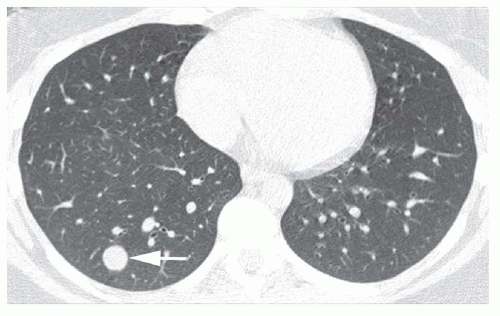
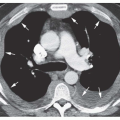
The lobe or segment distal to the bronchial obstruction usually remains aerated because of collateral ventilation
(
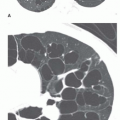
Figs. 1-4 and
1-5). In 90% of cases, air trapping in the distal lung results in decreased perfusion; radiographs and CT show the affected lung to be hyperlucent and hypovascular. Affected lung is often increased in volume, resulting in mediastinal shift or shift of a fissure. In 80% of cases, mucus accumulates within dilated bronchi distal to the obstruction, resulting in a tubular, branching, or ovoid density (mucous plug or
mucocele). Mucus within dilated bronchi usually appears low in attenuation. Expiratory radiographs or CT scans show air trapping (see
Fig. 1-5B).
The combination of these typical radiographic or CT findings in a young patient is strongly suggestive of the diagnosis. Bronchoscopy may be warranted to rule out another cause of bronchial obstruction, such as tumor.
CONGENITAL LOBAR EMPHYSEMA
CLE, also known as congenital lobar hyperinflation, is characterized by marked overinflation of a lobe (
Table 1-3). Most cases present within the first month of life; symptoms of respiratory distress are typical. Presentation after the first month may occur.
Most cases of CLE are associated with partial or complete bronchial obstruction occurring as a result of (a) deficient cartilage; (b) external compression, usually by an anomalous vessel or bronchogenic cyst; or (c) luminal abnormalities such as mucosal folds. Some cases are unassociated with bronchial obstruction.
CLE is most common in the left upper lobe, followed by the right middle lobe and right upper lobe. Only a few percent
occur in the lower lobes. Radiographs typically show marked overinflation and air trapping in the affected lobe. However, in neonates, the affected lobe may sometimes appear opaque because of retained fetal lung fluid. Mediastinal shift away from the abnormal lobe often occurs, and normal lobes are reduced in volume. Resection is often necessary.
It is reasonable to assume that cases of CLE that go unrecognized at birth may be diagnosed years later as bronchial atresia.
PULMONARY BRONCHOGENIC CYST
Bronchogenic cysts are foregut duplication cysts and result from abnormal development of the lung bud. They are lined by pseudostratified ciliated columnar epithelium, typical of bronchi. The cyst wall may also contain smooth muscle, mucous glands, or cartilage. Bronchogenic cysts are filled with fluid, which can be serous, hemorrhagic, or highly viscous and gelatinous because of its high protein content.
Bronchogenic cysts may be mediastinal or pulmonary. Mediastinal bronchogenic cysts are much more common than pulmonary cysts. They are discussed along with mediastinal masses in
Chapter 8.
Pulmonary bronchogenic cysts are most common in the medial lung and the lower lobes (
Table 1-4). They are sharply circumscribed and round or oval. The cyst wall may calcify. Rarely, the cyst may contain milk of calcium and appear dense. Cysts may slowly increase in size; a rapid increase in size is unusual unless infection occurs.
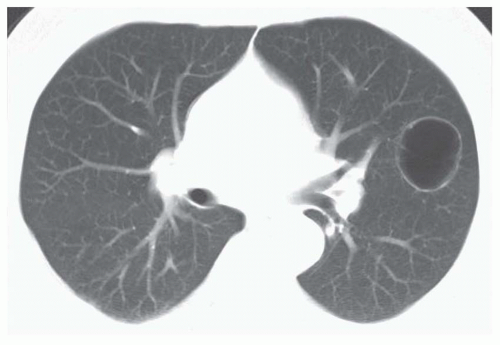
About half of fluid-filled bronchogenic cysts appear to be low in attenuation on CT (0 to 20 HU) (
Fig. 1-6). However, as with mediastinal bronchogenic cysts, the CT attenuation of a pulmonary bronchogenic cyst is variable. High CT numbers (40 to 80 HU), suggesting a solid mass, can be seen. Such cysts contain bloody or thick, proteinaceous fluid. Typically, the cyst wall appears very thin on CT or is invisible. A bronchogenic cyst may sometimes appear to be related to a small bronchus.
Infection eventually occurs in 75% of cases. In the presence of acute infection, a rapid increase in size of the cyst may be seen. Also, the outer cyst wall may become less well defined because of surrounding lung inflammation. During or after infection, a cyst may contain air (
Fig. 1-7; see also
Fig. 9-29 in
Chapter 9) or a combination of air and fluid (with an air-fluid level). When the cyst contains air, its wall appears very thin.
CONGENITAL CYSTIC ADENOMATOID MALFORMATION (CONGENITAL PULMONARY AIRWAY MALFORMATION)
Congenital cystic adenomatoid malformation (CCAM), also termed congenital pulmonary airway malformation (CPAM) consists of a multicystic, intralobar mass of disorganized lung tissue, derived primarily from bronchioles. About 70% present during the first week of life, but 10% are diagnosed after the first year, and rare cases in adults have been reported.
CCAM/CPAM can involve an entire lobe. Lower lobes are most often involved, but any lobe can be affected. The CCAM/CPAM communicates with the bronchial tree and is supplied by the pulmonary artery; systemic arterial supply is rarely present.
CCAMs are typically classified into three primary types (Types 1 to 3), which have different histology, gross pathologic findings, radiographic appearance, and prognosis (
Table 1-5).
Type 1 CCAMs (65% of cases) contain one or more cysts more than 2 cm in diameter (
Fig. 1-8). They usually appear radiographically as a large, air-filled multicystic lesion, sometimes with air-fluid levels, which may occupy the entire hemithorax.
Type 2 CCAMs (20% to 25% of cases) contain multiple cysts less than 2 cm in diameter. They present radiographically as an air-filled multicystic mass or a solid mass or area of consolidation (see
Fig. 1-9). This type may be associated with a poor prognosis because of associated renal and cardiac abnormalities.
Type 3 CCAMs (10% of cases) contain microscopic (less than 3 to 5 mm) cysts and present radiographically as a solid mass.
Two additional types of CCAM have been proposed, Types 0 and 4, based on histopathologic findings and the type of airway epithelium involved in the malformation. The term CPAM (congenital pulmonary airway malformation) has been suggested to encompass this expanded classification. Types 0 and 4 CCAM/CPAM each account for a few percent of cases. Type 0 CCAM/CPAM is associated with small cysts,
as in Type 3. Type 4 CCAM/CPAM is associated with large cysts, as is Type 1; its radiographic and CT appearance is the same as for Type 1 or 2.
Sonography can be used for prenatal diagnosis. Findings include polyhydramnios, fetal hydrops, and a solid or cystic mass in the fetal thorax. Fetal surgery may be attempted.
In infancy, CCAM/CPAM presents as a space-occupying lesion. Symptoms of respiratory distress are common. In neonates, CCAM/CPAM usually presents as a solid mass regardless of its type. Types 1 and 2 may become air filled over a period of days to weeks. They are often associated with progressive air trapping and mediastinal shift to the opposite side.
The treatment of choice is excision of the affected lobe. The prognosis of neonates with CCAM/CPAM is adversely affected by large size, underdevelopment of uninvolved lung (i.e., the presence of associated lung hypoplasia), and the presence of associated fetal hydrops or other congenital anomalies.
In adults, CCAM/CPAM usually presents as an air-filled or air- and fluid-filled cystic or multicystic mass. Most adults present with recurrent pneumonia, although recurrent pneumothorax has also been associated. Occasionally, bronchioloalveolar carcinoma may arise in relation to a CCAM/CPAM.
PULMONARY ARTERIOVENOUS MALFORMATION
Congenital arteriovenous malformation (AVM), also known as arteriovenous fistula, likely results from deficient formation or abnormal dilation of pulmonary capillaries due to a developmental defect in the capillary wall. From 35% to 67% of cases are associated with Osler-Weber-Rendu syndrome (
hereditary hemorrhagic telangiectasia), in which AVMs are found in the skin, mucous membranes, and viscera (
Table 1-6). Also, AVMs may rarely occur in patients with hepatopulmonary syndrome or as a result of trauma.
Pulmonary arteriovenous fistulas slowly enlarge over time and are usually first diagnosed in adulthood. More than two thirds of AVMs are found in the lower lobes, and they are typically subpleural in location. Fistulas are multiple in 35% of patients and bilateral in 10%.