- •
Compression of the heart, with small cardiothoracic ratio.
- •
Abnormal position of the heart within the chest.
- •
Distortion of the inferior vena cava as it enters the chest.
- •
Impaired filling of the heart can lead to findings on Doppler echocardiography of equilibration of peak E and A wave velocities and increased reversal of atrial contraction wave form in the inferior vena cava.
- •
Hydrops fetalis.
Anatomy and Anatomical Associations
Congenital cystic adenomatoid malformation (CCAM) is a rare developmental abnormality of the lungs. It is characterized as a benign hamartoma or dysplastic lung tumor due to overgrowth of terminal bronchioles. The tumor can grow to be quite large, compressing adjacent lung tissue and impairing growth of normal pulmonary tissue. CCAM can grow to become a large and significant size intrathoracic space-occupying lesion. Cardiovascular compromise as a result of compression of the heart and vascular structures can occur. When a critical size is reached, hydrops may develop that heralds impending fetal demise in nearly all cases, unless intervened upon. The majority of fetuses with CCAM have a relatively good outcome, but appropriate identification and ongoing surveillance are required because growth patterns can be unpredictable, with significant and rapid growth potential noted between 18 and 26 weeks of gestation.
CCAM is typically unilateral and usually involves only one lobe of the lung. Cystic structures arise from overgrowth of the terminal bronchioles with a commensurate reduction in number of alveoli. A variety of classifications have been described based on the histopathology; however, our group at Children’s Hospital of Philadelphia (CHOP) have found it most useful to characterize them in two ways based on gross anatomy and ultrasound findings. Macrocystic lesions contain single or multiple cysts that are 5 mm in diameter or larger on prenatal ultrasound, whereas microcystic lesions appear as a solid echogenic mass. In the macrocystic type, when multiple cysts are present, they typically communicate with one another.
CCAM is to be distinguished from other types of intrathoracic lung lesions in the fetus, in particular bronchopulmonary sequestration (BPS). A BPS is a mass of nonfunctioning lung tissue that is supplied by an anomalous systemic artery and does not have a bronchial connection to the native tracheobronchial tree. On ultrasound, BPS appears as a well-defined echo-dense homogeneous mass and can be confused with a microcystic CCAM, except that the former has an arterial supply from the systemic arterial system. In CCAM, the arterial supply is from the pulmonary arterial tree and venous drainage is through the pulmonary veins. In BPS, the arterial supply is typically from a large artery emanating from the descending aorta and the venous drainage is through the pulmonary venous system. Hybrid lesions, with characteristics of both BPS and CCAM, have been described. Both can cause significant cardiovascular compromise when they grow to a large size. Other lesions to be distinguished include congenital lobar emphysema and peripheral bronchial atresia. Occasionally, congenital diaphragmatic hernia can be confused with CCAM. Careful ultrasound or ultrafast magnetic resonance imaging (MRI) evaluation should be able to distinguish these very different anomalies with different management schema and outcome.
Frequency, Genetics, and Development
The incidence of CCAM in the general population is not known because small lesions may be subclinical and some lesions become smaller and resolve during in utero development or early childhood.
CCAM is believed to occur because of an abnormality of lung development. The development of the mammalian vertebrate lung is divided into five distinct periods based on the anatomical changes that take place in lung architecture: embryonic (3-7 wk), pseudoglandular (7-17 wk), canalicular (17-29 wk), saccular (24-36 wk), and alveolar (36 wk to maturity). In the pseudoglandular period, there is rapid expansion of the conducting airways and peripheral lung tubules, which continue to branch and bud to form acinar tubules. The expansion of these small tubules in the periphery of the lung produces a glandular appearance. Uncontrolled growth and development of the macrocystic CCAM is believed to occur during the pseudoglandular phase, whereas microcystic CCAM is believed to develop later at the canalicular phase. Eight to 10 weeks of gestation is reported to be a particularly vulnerable period of time for teratogen impact on lung development leading to CCAM.
Resected large CCAM specimens demonstrate increased cell proliferation and markedly decreased apoptosis when compared with gestational age–matched normal fetal lung tissue. Factors that enhance cell proliferation or down-regulate apoptosis have been studied including keratinocyte growth factor, platelet-derived growth factor, and fibroblast growth factor (FGF). Using a rat transgenic lung model, investigators have shown that FGF-10 overexpression in the proximal tracheobronchial tree during the pseudoglandular phase of lung development resulted in large cysts, whereas FGF-10 overexpression in the distal lung parenchyma during the canalicular phase resulted in small cysts, providing a possible developmental model for the anomaly.
We reported on the possible association of CCAM and congenital heart disease when in the presence of genetic abnormality. Of 262 fetuses evaluated with CCAM at our center between 2000 and 2006, 4 (1.6%) had associated congenital heart disease consisting of 2 with ventricular septal defect, 1 with tetralogy of Fallot, and 1 with transposition of the great arteries. A review of the literature reveals that nearly two thirds of fetuses with CCAM and congenital heart disease have a known genetic/chromosomal abnormality.
Prenatal Physiology
Large lung lesions such as CCAM have predictable pathophysiological effects on the fetus. Esophageal compression by the mass causes altered swallowing of amniotic fluid and results in polyhydramnios. Most important, a large mass can distort intrathoracic structures and compress the heart.
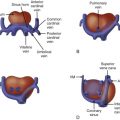
The fetal myocardium normally exists in a relatively noncompliant “stiff” state, with limited ability to increase preload or stroke volume. Extrinsic compression by a large intrathoracic, space-occupying lesion such as a CCAM can quickly lead to underfilling of the fetal heart, limiting cardiac output ( Figure 43-1 ). Compression or tamponade of the fetal heart leads to increased atrial pressure and increased venous pressure, resulting in hydrops. The heart can appear “squeezed” within the chest cavity and is markedly distorted and deviated away from the side of the CCAM mass. While the heart is pushed over, the abdominal contents remain relatively fixed and the inferior vena cava can become distorted in its course through the abdomen, as it stretches to reach the abnormally positioned right atrium ( Figure 43-2 ). Such inferior vena caval distortion may limit venous return, further contributing to the development of ascites. Interestingly, increased intrathoracic pressure and tension created by the CCAM mass inhibits the formation of pleural or pericardial effusion.


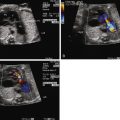
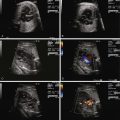
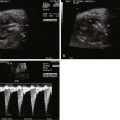
In order to obtain a clearer sense of the cardiovascular physiology in CCAM lesions, we investigated the echocardiographic and Doppler characteristics that distinguish the fetal CCAM with and without hydrops. In a series of 41 fetuses with CCAM, hydrops was present in 15 (37%). Those that developed hydrops had a lower cardiothoracic ratio than those without hydrops, although in both, the heart size was smaller than normal (0.18 vs. 0.23, P = 0.001). The fetuses with hydrops also demonstrated increased early diastolic filling, with the ratio of early filling to atrial contraction (E/A ratio) significantly higher for both tricuspid and mitral valves. In addition, the degree of reversal of flow with atrial contraction in the inferior vena cava as well as frequency of presence of umbilical venous pulsation was higher in the hydrops group. Overall systolic performance of both the right and the left ventricle appeared to be normal or hyperdynamic. These findings are consistent with a picture of impaired filling and cardiac tamponade.
In another study, we looked at echocardiographic Doppler-derived parameters of ventricular performance in 36 fetuses with CCAM at 25 weeks’ gestation and compared them with age-matched controls. We studied the myocardial performance index (MPI), a global measure of systolic and diastolic performance, the ejection force, a preload-dependent measure of systolic performance, and cardiac output as an overall measure of cardiovascular productivity, because cardiac output/blood flow is the end result of cardiac performance ( Table 43-1 ). For the fetuses with CCAM, MPI values were higher than normal, suggesting worse global dysfunction, and ejection force was lower than normal, suggesting a decrease in the volume of ejection. Combined cardiac output was approximately 17% lower in the CCAM group relative to normal.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


