- •
Displacement of the cardiac position.
- •
Small left ventricle cavity volume.
- •
Assess mitral valve annulus and aortic annulus, which may be of normal size despite small left ventricle cavity volume.
- •
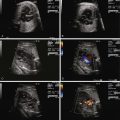
Assess aortic arch dimensions and direction of flow.
- •
Heart function is typically normal.
Anatomy and Anatomical Associations
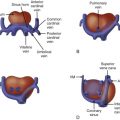
Congenital diaphragmatic hernia (CDH) is a defect in the muscular diaphragm separating the chest and abdomen. Extrusion of abdominal contents into the thoracic cavity leads to pulmonary hypoplasia and pulmonary hypertension after birth. CDH can be classified anatomically as Bochdalek or Morgagni type. Bochdalek hernia is most common and is a defect located in the posterolateral aspect of the diaphragm. The majority of Bochdalek hernias (85%) occur on the left side, some on the right side, and occasionally, it can be bilateral. Because the liver is situated on the right, it is deemed to be somewhat protective during development. Morgagni hernias are retrosternal or parasternal diaphragmatic defects and make up 2% of cases.
CDH is often associated with other anomalies and syndromes, congenital heart disease being quite common. Congenital heart disease occurs in 11% to 15% of CDH cases without a recognizable genetic syndrome, but the incidence may be even higher when syndromes are included. The spectrum of heart disease mimics what is seen in the general population, ventricular septal defect being most common. There are some reports of a slightly increased frequency of conotruncal defects with CDH as well as left-sided obstructive disease; however, these descriptions do not separate out those in which CDH is present within the context of a syndrome that may also favor a particular type of heart defect.
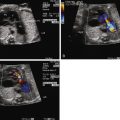
CDH is associated with a number of structural abnormalities of the fetal heart. Myocardial mass in CDH is smaller than normal. Left ventricular dimensions, in particular cavity size and wall thickness, are smaller than normal. Left ventricular “hypoplasia” has been described; however, left ventricular smallness in CDH is quite possibly a consequence of an underfilled ventricular cavity than true hypoplasia of the cavity due to abnormal development, because the mitral and aortic valve annulus diameters are typically within range of normal limits for gestational age or only slightly smaller. However, there are conflicting data on this matter.
The most important impact of CDH is on development of lung hypoplasia and pulmonary vascular disease. This feature dictates the clinical severity of the disease and its management.
Frequency, Genetics, and Development
CDH occurs in 1 of every 2000 to 4000 live births and makes up approximately 8% of all birth defects. Prenatal ultrasound has increased the prevalence of CDH, because it is now possible to capture many severe cases for which intensive perinatal management can be offered and in which survival would previously have been unlikely.
Formation of the diaphragm and separation between the chest and the abdomen takes place between the third and eighth week of gestation. CDH occurs as a defect in muscle formation, or fusion, of the various components that contribute to the diaphragm. If the diaphragm is incomplete, the intestines, stomach and liver can migrate into the chest cavity during critical times of lung development, leading to pulmonary hypoplasia. The effect is greatest on the side of the herniation; however, forces may impair normal development of the contralateral lung as well.
Bowel herniation into the chest cavity during critical periods of lung development results in inadequate formation of the bronchial tree, limiting airway divisions. Normally, airway development results in 23 to 35 generations of airway division, whereas in CDH only 12 to 14 develop on the affected side and 16 to 18 on the contralateral side. Alveolarization is reduced as well and there is functional deficiency in the surfactant and antioxidant systems. Pulmonary vascular development parallels airway development. CDH leads to abnormal formation of the pulmonary arterioles with increased muscularization, with far fewer arterial branches and truncated arborization.
Although most of the structural abnormalities seen in CDH are presumed related to mechanical factors due to bowel contents in the thorax, some data suggest the presence of intrinsic abnormalities of the myocardium. A nitrofen-induced rat model for CDH has been extensively studied. Investigators have shown evidence for thinning of the myocardium and left ventricular smallness with down-regulation of various growth factors, independent of the degree of mechanical displacement. Alterations in extracellular matrix and myocardial immaturity have also been described. These studies suggest that an associated inherent developmental abnormality of the myocardium may exist in the fetus with CDH.
Whereas a number of syndromes are associated with CDH, a few in particular are highly associated with congenital heart disease and are listed in Table 44-1 .
| SYNDROME | FREQUENCY OF CONGENITAL HEART DISEASE | TYPE |
|---|---|---|
| Wolf-Hirschhorn syndrome (deletion 4p) | 30-50% | ASD secundum, PS, VSD |
| Trisomy 18 | 95% | VSD, TOF, DORV, polyvalvar dysplasia |
| Brachmann-de Lange syndrome | 25% | ASD, VSD, PS |
| Simpson-Golabi-Behmel syndrome | 26% | No specific type |
| Fryns’ syndrome | 50% | ASD, VSD, conotruncal anomalies |
| Thoracoabdominal syndrome | 50% | No specific type |
| Teratogen exposure—vitamin A | 45% | VSD, defects of aorticopulmonary septation, aortic anomalies |
| Teratogen exposure—nitrofen (rats) | 60% | Conotruncal anomalies, outflow tract anomalies, aortic arch anomalies |
Prenatal Physiology
CDH affects fetal cardiovascular physiology in a variety of important ways. Because the most common location for CDH is on the left, abdominal contents in the chest cavity enter the left chest and can cause mechanical compression of the heart, reducing the potential for ventricular filling. Distortion of the position of the entry site of the inferior vena cava (IVC) and ductus venosus (DV) into the floor of the right atrium due to cardiac compression can alter intracardiac flow patterns. Normally, IVC and DV flows are directed toward the foramen ovale which fill the left ventricle with relatively highly oxygenated blood in order to perfuse the essential organs of the heart and brain. In CDH, such streaming may be adversely affected owing to torsion of the heart and alteration in axis, with the IVC and DV angled away from the foramen ovale. This will limit the filling volume of the left ventricle but increase filling of the right ventricle. Enlargement of the right ventricle may further compress the left or give rise to the impression of relative size discrepancy between the two ventricles. Furthermore, pulmonary hypoplasia implies a decrease in pulmonary vascularity. Hence, pulmonary venous return is diminished in CDH because there is less lung to perfuse, further contributing to a decrease in left ventricular filling volume. Chronic underfilling and decreased preload to the left ventricle contribute to the relatively smaller left ventricular cavity size and thinner myocardial walls seen in CDH.
Chronic impaired filling probably plays a more important role in contributing to left ventricular smallness in CDH than actual pressure-induced mechanical compression. First, there is a general relationship between the degree of pulmonary hypoplasia and the degree of myocardial mass diminution in the animal model. Second, left ventricular hypoplasia is not seen in cases of congenital cystic adenomatoid malformation or bronchopulmonary sequestration, lesions in which intrathoracic tension is quite high. In fact, in these anomalies, pulmonary venous return is greater than normal, which further supports the notion that it is volume depletion and not increased pressure that is the predominant factor leading to left ventricular changes in CDH.
Prenatal Management
Ultrasound and magnetic resonance imaging (MRI) evaluation are helpful in planning a management strategy and in stratifying risk in the fetus with CDH. Assessment of the location of the defect and identification of the position of the organs are important. The position of the liver is critical, because those fetuses with the liver “up” in the chest have greater likelihood for pulmonary hypoplasia and have a poorer prognosis. Determination of the liver position can be made by ultrasound; however, fetal MRI provides a clearer image of the relationship of the liver to adjacent lung tissue, which may appear of similar echodensity on ultrasound.
The “lung-head ratio” (LHR) is an index for measuring the degree of lung hypoplasia present and is determined by calculating the lung volume present on the side of the defect indexed to the head circumference. Controversy exists as to the best way to calculate the LHR and for its prognostic value; however, many have used it as a general way of gauging the severity of disease. The lower the LHR the worse the pulmonary hypoplasia and prognosis. A review of our experience at Children’s Hospital of Philadelphia (CHOP) with 89 fetuses found that a low LHR of less than 1.0 predicted increased likelihood of needing extracorporeal membrane oxygenation (ECMO) support (75%) and poorer survival (35%); however, when LHR was measured at less than 24 weeks’ gestation, it was much less predictive. Liver position was a much better predictor of outcome. Whereas liver “up” fetuses required ECMO support in 80% of cases and survival was 45%, survival for those with liver “down” exceeded 90%.
Cardiovascular evaluation via fetal echocardiography is indicated in all fetuses with CDH, because the presence of significant congenital heart disease is common and seriously affects survival. When no congenital heart disease is present, echocardiography should focus on the degree of heart distortion within the chest and the change in cardiac axis. Left ventricular dimensions should be measured with particular attention to the mitral and aortic annulus diameters, because if these are normal, the left ventricular cavity size is likely to be normal as well (when there is an intact ventricular septum, as the mitral valve goes . . . so goes the ventricle!). The aortic arch can oftentimes appear slightly small in CDH and its course is distorted in the mediastinum, making it difficult to identify the presence of a coarctation of the aorta. Measurements of the isthmus may be helpful; however, confident determination of arch anatomy in CDH, and ruling out a coarctation of the aorta when arch structures appear small, must oftentimes await postnatal evaluation.
Much effort has been exerted in finding an appropriate in utero treatment strategy for CDH. Early efforts at open fetal surgery with direct CDH repair yielded results that were no better than conventional postnatal management. With improvements in postnatal care and neonatal outcome, fetal intervention for CDH is reserved for those with the worst prospects for good outcome.
The notion of increasing airway pressure to mechanically induce lung growth and development is currently of great interest. It has been argued that increased airway pressure can actually result in growth of normal lung tissue and vasculature. Initially, this was achieved through open techniques utilizing tracheal clips; however, currently, this can be achieved through a laparoscopic approach using a detachable balloon. The benefits of such a fetal endoscopic tracheal occlusion (FETO) procedure include (1) the lack of need for open uterine incision and its potential consequences of premature labor, (2) it is a relatively quick procedure performed under local anesthetic, and (3) there is the ability to percutaneously deflate the balloon before delivery. FETO has been demonstrated to be an effective strategy when undertaken by experienced hands in select cases.
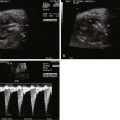
Investigators have looked at cardiac dimensions and measures of performance in CDH. As expected, in comparison with normal matched controls, end-diastolic and end-systolic dimensions were smaller by 32% and 37%, respectively. However, measures of ventricular systolic and diastolic function including ejection fraction, shortening fraction, ratio of early filling (E wave) to atrial filling (A wave) velocities, and the myocardial performance index (MPI) were no different from normal.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


