Fig. 1.1
Classification System of Cardiomyopathies proposed by the European Society of Cardiology [4]. ARVC arrhythmogenic right ventricular cardiomyopathy, DCM dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, RCM restrictive cardiomyopathy
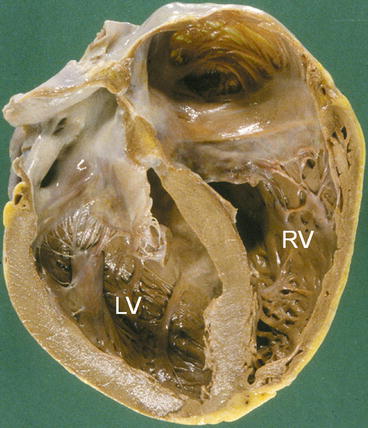
Fig. 1.2
Gross pathologic specimen of a case of Dilated cardiomyopathy who died suddenly. Left ventricle (LV) is grossly enlarged, without evident wall hypertrophy. RV right ventricle
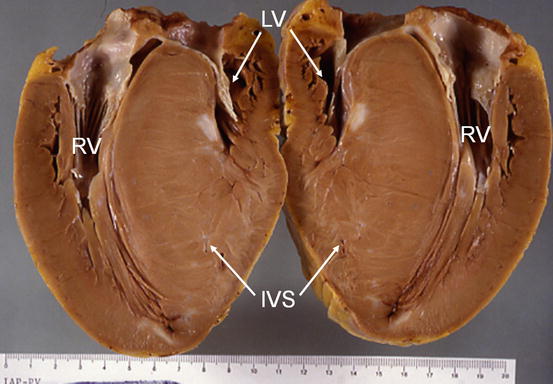
Fig. 1.3
Case of Hypertrophic cardiomyopathy. Severe left ventricular (LV) hypertrophy, predominant at the level of interventricular septum (IVS) is evident. LV chamber is small. The right ventricle (RV) is also hypertrophic
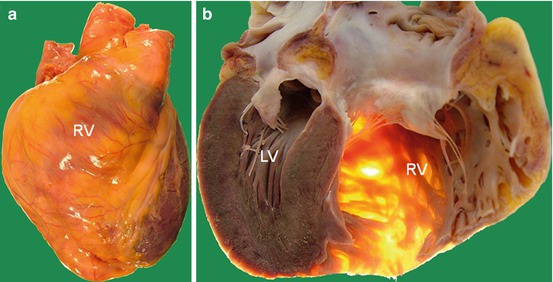
Fig. 1.4
Case of arrhythmogenic right ventricular cardiomyopathy. (a): External appearance of anterior aspect of the heart shows an enlarged right ventricle (RV) with a yellowish appearance, compatible with fatty infiltration. (b): section of the heart (4 chamber view) confirms the severe RV dilatation and shows the extreme wall thinning of RV (note the transillumination of RV thinned wall). LV left ventricle
Very recently the World Heart Federation published a new comprehensive classification, the so called MOGE(S) classification [6]. This descriptive nosologic system was constructed in a similar manner as the TNM system universally used for staging tumors. Each cardiomyopathy can be defined according to 5 characteristics: M describes the morphofunctional pattern of the CMP, such as HCM, DCM, etc; O the organ involvement; G the genetic/familial inheritance pattern, and E an explicit etiological annotation with details of genetic defect or underlying disease/cause: in addition S as an option describes the functional status using the ACC/AHA stage and NYHA functional class. The MOGE(S) classification can be considered a compromise between the American [3] and European [4] classifications and is expected to be a major advance in the field of CMPs, allowing, as stated by the Authors, “better understanding of the disease, easier communication among physicians and helping to develop multicenter/multinational registries to promote research in diagnosis and management of cardiomyopathies”.
1.3 Clinical Relevance
Exceptional progresses of knowledge in the field of CMPs have been done in the last decades. However, despite many causing disease genetic mutations have been found, and some specific or common pathophysiologic pathways have been hypothesized, controversial and open issues are still present that should be faced by future basic and clinical research.
Some important points of clinical relevance in CMPs have to be mentioned, as the role of familial/genetic screening, and the importance of systematic follow-up and specific registries.
The ongoing evidence of the relevance and frequency of genetic CMPs [7] should indicate a systematic familial screening in all 1st degree family members of probands. In fact, familial screening can provide an earlier diagnosis with a subsequent better long-term outcome [8].
Moreover, patients affected by CMPs require a regular long-term follow-up for continuous monitoring of the evolution of disease, better assessment of the effects of treatment, and risk re-stratification [9]. An important point to remember is that the familial and genetic screenings and the regular long-term follow-up have important costs in terms of human and economic resources. CMPs also set important ethical issues – such as the implications of the identification at genetic screening of apparently non affected family members with presence of gene mutation [7], potential risk of pregnancy, and employment or sport activity in young patients without other associated diseases – that the clinical cardiologist has to face often without specific guidelines. Thus, in absence of specific clinical trials, the presence of registries enrolling clinical, instrumental and prognostic data of large cohorts of patients affected by CMPs and systematically followed in the long-term has a paramount relevance for the correct management of these diseases by clinical cardiologists. In Tables 1.1 and 1.2 are respectively reported the recruitment rate and the summary of enrolled patients and length and number of follow-up evaluation of the Heart Muscle Disease Registry of Trieste, active from 1978.
 Dilated Cardiomyopathy: Usefulness of Imaging in Prognostic Stratification and Choice of Treatment
Dilated Cardiomyopathy: Usefulness of Imaging in Prognostic Stratification and Choice of Treatment
 Hypertrophic Cardiomyopathy: Clinical Assessment and Differential Diagnosis
Hypertrophic Cardiomyopathy: Clinical Assessment and Differential Diagnosis
 Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Assessment and Differential Diagnosis
Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Assessment and Differential Diagnosis
 Advanced Echocardiographic Technologies in Dilated Cardiomyopathy
Advanced Echocardiographic Technologies in Dilated Cardiomyopathy
 Infiltrative/Storage Cardiomyopathies: Clinical Assessment and Imaging in Diagnosis and Patient Management
Infiltrative/Storage Cardiomyopathies: Clinical Assessment and Imaging in Diagnosis and Patient Management
 Restrictive Cardiomyopathy: Clinical Assessment and Imaging in Diagnosis and Patient Management
Restrictive Cardiomyopathy: Clinical Assessment and Imaging in Diagnosis and Patient Management




Table 1.1
Recruitment rate of patients in Heart Muscle Disease Registry of Trieste (1978–31/12/2013)
Disease | |||||
|---|---|---|---|---|---|
Year of recruitment | IDCM | Myocarditis | ARVC | HCM | Total |
Before 1979 | 5 | 2 | 7 | ||
1979–1980 | 17 | 1 | 18 | ||
1981–1982 | 10 | 2 | 3 | 15 | |
1983–1984 | 20 | 15 | 7 | 1 | 43 |
1985–1986 | 38 | 17 | 6 | 6 | 67 |
1987–1988 | 41 | 4 | 8 | 12 | 65 |
1989–1990 | 60 | 4 | 5 | 13 | 82 |
1991–1992 | 80 | 4 | 7 | 7 | 98 |
1993–1994 | 99 | 7 | 13 | 22 | 141 |
1995–1996 | 89 | 7 | 10 | 16 | 122 |
1997–1998 | 62 | 3 | 6 | 10 | 81 |
1999–2000 | 74 | 2 | 5 | 12 | 93 |
2001–2002 | 69 | 2 | 5 | 8 | 84 |
2003–2004 | 71 | 4 | 7 | 11 | 93 |
2005–2006 | 80 | 9 | 4 | 22 | 115 |
2007–2008 | 75 | 7 | 15 | 45 | 142 |
2009–2010 | 51 | 8 | 7 | 43 | 109 |
2011–2013 | 90 | 11 | 8 | 48 | 157 |
Total n° of patients | 1,031 | 106 | 119 | 276 | 1,532 |
Related posts:
Dilated Cardiomyopathy: Usefulness of Imaging in Prognostic Stratification and Choice of Treatment
Hypertrophic Cardiomyopathy: Clinical Assessment and Differential Diagnosis
Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Assessment and Differential Diagnosis
Advanced Echocardiographic Technologies in Dilated Cardiomyopathy
Infiltrative/Storage Cardiomyopathies: Clinical Assessment and Imaging in Diagnosis and Patient Management
Restrictive Cardiomyopathy: Clinical Assessment and Imaging in Diagnosis and Patient Management
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
