Disease
Echocardiographic features
Infiltrative cardiomyopathies
Amyloidosis
Left and right ventricle hypertrophy with preserved ventricular size
Granular left ventricle appearance
Biatrial dilatation
Thickened interatrial septum and valve leaflets
Pericardial effusion
Restrictive physiology
Decreased ejection fraction in advanced cases
Sarcoidosis
Variable wall thickness
Normal to dilated ventricular chambers
Scar retraction, aneurysms in the left ventricle
Focal or global hypokinesis
Pulmonary hypertension
Wegener granulomatosis
Wall motion abnormalities
Pericardial effusion
Storage cardiomyopathies
Fabry disease
Predominant concentric hypertrophy of left ventricle
Right ventricular and papillary muscle hypertrophy also common
Mild valve abnormalities
Mild dilatation of the aortic root
Possible binary appearance of the endocardial border
Danon disease
Marked symmetrical increase in left ventricular wall thickness (range 20–60 mm)
PRKAG2 cardiac syndrome
Left ventricle hypertrophy ranging between asymmetric septal hypertrophy, concentric hypertrophy, and distal hypertrophy
Glycogenoses
Pompe disease: severe thickening of the intraventricular septum, free wall, and posterior left ventricular wall, with a tumor-like appearance of the papillary muscles
Cori-Forbes disease: concentric left ventricle thickening, increased refractile pattern, and granular appearance of the myocardium
Friedreich ataxia
Increase in left ventricular septal and posterior wall thickness
Cavity size and ejection fraction usually normal
Oxalosis
Biventricular symmetrically thickened walls
Myocardium characterized by patchy, echo-dense speckled reflection
Mucopolysaccharidoses
Asymmetrical septal hypertrophy
Thickening of the valves
Mitral and the aortic valves with insufficiency and/or stenosis
Hemochromatosis
Increase in left and right wall thickness
Dilated left ventricle with global systolic dysfunction
Restrictive physiology
Mitochondrial cardiomyopathy
Left ventricular hypertrophy
In the late stage of disease, left ventricular dilatation and systolic dysfunction (hypokinetic end-stage evolution)
Table 20.2
Role of different imaging techniques in the assessment of infiltrative/storage cardiomyopathies
Echocardiography | CMR | PET | SPECT | CT | |
|---|---|---|---|---|---|
Cardiac amyloidosis | ++ | +++ | + | ++ | |
Anderson-Fabry disease | ++ | +++ | + | + | |
Cardiac hemochromatosis | + | +++ | + | ||
Cardiac sarcoidosis | + | +++ | +++ | ++ |
Heart failure (HF) is a common finding in infiltrative/storage CMP. However, as HF represents the final clinical pathway of many cardiac diseases, the recognition of many other multi-system manifestations (e.g., renal failure, peripheral neuropathy, skeletal myopathy, mental retardation, hepatosplenomegaly, etc.) is crucial to correctly direct the diagnosis [1] (Table 20.3). Moreover, it is important to emphasize that, especially in infiltrative CMP, evidence of a left ventricular (LV) hypertrophy on electrocardiography (ECG) is not a reliable indicator of increased LV mass. Indeed, infiltrative disorders mainly cause increased wall thickness by accumulation of abnormal substances in the interstitium and not within the myocytes (pseudohypertrophy). Consequently, increased wall thickness does not consistently correlate with high QRS complex voltages at ECG, and paradoxical low voltages or normal QRS voltages at ECG, contrasting with the significant LV hypertrophy, can be the first diagnostic clue of infiltrative CMP (typically amyloidosis) [1]. On the other hand, storage CMP, characterized by intracellular myocardial accumulation, frequently show very high QRS voltages at ECG, sometimes associated with ventricular pre-excitation, which can be the ECG hallmark of these disorders (e.g., Fabry disease, Danon disease).
Table 20.3
Infiltrative and storage cardiomyopathies (CMP): clinical features and electrocardiograph (ECG) profile
Age at presentation | Incidence/prevalence | ECG profile | Clinical features | |
|---|---|---|---|---|
Cardiac amyloidosis | >30 years | Depending on amyloidosis subtype | Low QRS voltages, pseudoinfarction pattern in anterior leads, AV and IV blocks | Multiple myeloma, dysphonia, macroglossia, skin alterations, peripheral neuropathy, carpal tunnel syndrome, renal failure with proteinuria |
Cardiac sarcoidosis | >30–40 years | 10.9/100,000 population | AV blocks, IV conduction defects, VT, SVT | Multiple granulomas (lungs, lymphatic system, liver, skin, eyes, nervous system) |
Wegener granulomatosis | 45 years (infantile and senile forms have been described) | 2–12/1,000,000 live births | Aspecific | Necrotizing vascular granulomas involving multiple organs. Upper and lower airways chronic inflammation. Skin lesions, peripheral neuropathy, ocular abnormalities |
Primary cardiac lymphoma | >40 years | Rare (<1/1,000,000 population/year) | Low voltages, AV blocks | Usually affects immunocompromised patients. Paraneoplastic syndrome, cardioembolic strokes |
Fabry disease | Male: childhood/adolescence | 1/100,000 live births/year (probably underestimated) | LVH (++), ventricular pre-excitation | Angiokeratomas, acroparesthesias, pain crises, fever, fatigue, hearing loss, juvenile TIA/ictus, renal failure |
Female: late adolescence/adulthood | Can develop AV or IV conduction defects | |||
Danon disease | Male: ~16 years | Rare (<1/1,000,000) | LVH (++), ventricular pre-excitation/WPW syndrome | Skeletal myopathy (proximal), mental retardation, optical atrophy and retinal dysfunction, visual impairment |
Female: ~30 years | May present VT or SVT | |||
PRKAG2-gene-related CMP | >30 years | Unknown | LVH (++), ventricular pre-excitation/WPW syndrome. May present conduction abnormalities | May be associated with skeletal muscle glycogenosis |
Glycogenoses | 0–1 years | 1/100,000 live births | LVH (++), ventricular pre-excitation, conduction defects | Skeletal myopathy with hypotonia and elevated CK levels, hepatomegaly, metabolic disorders |
Friedreich ataxia | 10–15 years | 1/100,000 live births | Normal QRS voltages or mild LVH/VT. | Progressive gait ataxia followed by slurred speech and upper-limb ataxia; optic atrophy, hearing loss |
Cardiac oxalosis | From infancy (severe forms) to adulthood (mild forms) | 1/100,000 live births | AV blocks | Nephrocalcinosis, progressive renal failure, vascular calcifications, visual impairment |
Mucopolysaccharidoses | <10 years | 1/100,000 live births | May present low QRS voltages. Sudden death | Globally: mental retardation, coarse facies, skeletal abnormalities, macrocephaly, hypertrichosis, hepatosplenomegaly, upper airways obstructions, visual impairment, hearing loss |
Heart: valvular thickening, mitral prolapse, coronary artery disease, myocardial hypertrophy | ||||
Mucolipidoses | <10 years | Rare (<1/1,000,000) | See MPS | See MPS |
Gangliosidoses | <10 years | 1/200,000 live births | See MPS | See MPS |
Hemochromatosis | >30 years | Depending on hemochromatosis subtype | Normal QRS; can develop low voltages and repolarization abnormalities in advanced stages | Liver cirrhosis, diabetes mellitus, hypogonadotropic hypogonadism, arthritis |
Wilson disease | 3–50 years | 1/100,000 live births | LVH or biventricular hypertrophy, early repolarization, ST depression and T inversion | Liver cirrhosis, neurologic disorders (movement disorders or rigid dystonia), psychiatric manifestations (depression, phobias, compulsive behaviors, aggression or antisocial behavior, intellectual deterioration), Kayser-Fleischer rings (result from copper deposition in Descemet’s membrane of the cornea) |
Mitochondrial diseases (CPEO, Kearns-Sayre syndrome, Leigh syndrome, LHON, MELAS, MERRF, NARP, Pearson’s marrow-pancreas syndrome) | Variable | 9.2/100,000 adults aged <65 years | Ventricular pre-excitation/WPW syndrome, conduction disorders (AV blocks) | Encephalopathy, stroke-like episodes, seizures, cognitive impairment, depression, ataxia, sensorineural hearing loss, skeletal myopathy, palpebral ptosis, external ophthalmoplegia, optic atrophy, diabetes mellitus, hypothyroidism, hypogonadism, hypoparathyroidism, hepatic failure, renal tubulopathy |
Echocardiographic features are quite similar in most of infiltrative and storage CMP [2] (Table 20.1), frequently mimicking hypertrophic cardiomyopathy (HCM) in morphologic abnormalities and restrictive cardiomyopathy (RCM) from the hemodynamic point of view.
A final important point is that the use of advanced echocardiographic techniques, as well as CMR (Table 20.2), can be helpful in differential diagnosis, identifying early cardiac involvement, and quantifying the severity of cardiac damage in systemic infiltrative or storage diseases, with a favorable effect on treatment and outcome.
20.2 Infiltrative Cardiomyopathies
20.2.1 Cardiac Amyloidosis
Cardiac amyloidosis (CA) can be considered the prototype of infiltrative CMP. It results from pathological accumulation of insoluble extracellular fibrils derived by an abnormal folding of heterogeneous proteins [3] (Table 20.4). Regardless of which precursor protein causes the disease, the deposits are indistinguishable with light microscopy and invariably stain with Congo red staining (Fig. 20.2). The spectrum of organ involvement can include kidneys, blood vessels, nervous system, liver, lungs, gastrointestinal system, skin, eyes, joints, bones, and the heart.
Table 20.4
Amyloidosis subtypes
Type | Precursor protein | Systems/organs involved | Notes |
|---|---|---|---|
Primary amyloidosis (AL) | Immunoglobulin light chains | Heart, kidney, liver, gastrointestinal system, peripheral nervous system, autonomic nervous system | Most common form in Western countries. Associated with multiple myeloma or lymphoma. |
λ (75 %) κ (25 %) | |||
Secondary amyloidosis (AA) | Serum amyloid A protein | Kidney, autonomic nervous system, heart (uncommon) | Chronic inflammatory disorders |
Senile systemic amyloidosis (SSA) | Transthyretin (wild-type) | Heart | Advanced age (>80 years). Slowly progressive course, relatively benign prognosis. |
Hereditary systemic amyloidosis (ATTR, AApoA1) | Mutated transthyretin, mutated apolipoprotein A1 (or other rare variants) | Heart, kidney, nervous system | Autosomal dominant inheritance |
Atrial isolated amyloidosis | Atrial natriuretic peptide | Heart (atrium 100 %) | Increased risk of atrial fibrillation |
Dialysis-related amyloidosis (Aβ2M) | β2-microglobulin | Osteoarticular system | Long-lasting hemodialysis |
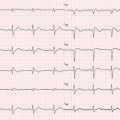
Cardiac involvement is common and represents the most frequent cause of morbidity and mortality [4]. Symptoms include HF (dyspnea, peripheral edema, ascites), angina, hypotension, syncope, and palpitations. Supraventricular arrhythmias, especially atrial fibrillation (AF), can occur in 10–15 % of cases [3]; typical ECG pattern includes (Fig. 20.1):
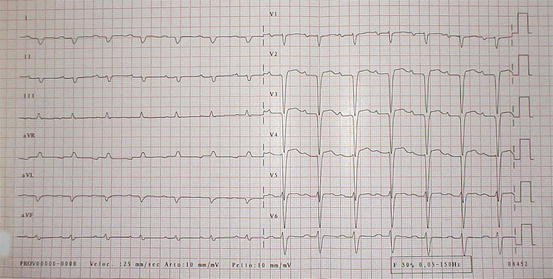
Fig. 20.1
Typical electrocardiogram (ECG) in a patient with cardiac amyloidosis. Low QRS voltages in peripheral leads and marked axis deviation are evident. Pathologic Q waves are also present
Low QRS voltages (46 % of cases)
Pseudonecrosis Q waves in anterior leads (64 %)
Abnormal QRS axis deviation (66 %)
Atrioventricular blocks (35 %)
Intraventricular blocks (16 %)
AF or flutter (20 %)
The presence of coexisting systemic manifestations, such as a multiple myeloma, dysphonia, macroglossia, skin alterations, peripheral neuropathy, carpal tunnel syndrome, renal failure, and proteinuria, can strengthen the suspicion of systemic amyloidosis.
CA is usually indirectly diagnosed using noninvasive cardiac imaging and a bioptic confirmation of amyloid presence in an extracardiac tissue (typically abdominal fat, rectal submucosa, or salivary glands). Endomyocardial biopsy (EMB) (Fig. 20.2) may be necessary in cases with a high clinical suspicion but nondiagnostic extracardiac histology [4]. Clinical phenotype, prognosis, and treatment are highly variable, depending on the type of amyloidosis (Table 20.4). The most common form is amyloid light-chain (AL) amyloidosis, which has an estimated incidence of six to ten cases per million population per year [5]. The precursor protein is an immunoglobulin light-chain (λ or κ) and is sometimes related to multiple myeloma or lymphoma. Median survival of patients with AL amyloidosis is around 48 months [6], and the presence of symptomatic cardiac involvement at diagnosis represents a significantly negative prognostic marker [7]. Other forms include hereditary amyloidosis [transthyretin (ATTR), apolipoprotein A1 (AApo A1)], senile systemic amyloidosis (SSA), secondary amyloidosis (AA), dialysis-related amyloidosis [β2‐microglobulin‐derived (Aβ2M)] and isolated atrial amyloidosis (for details, see Table 20.4) [8].
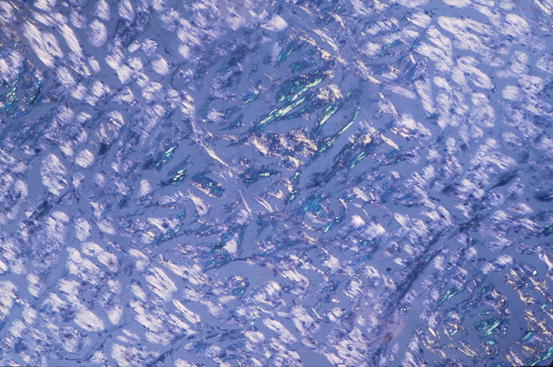
Fig. 20.2
Typical histologic pattern of cardiac amyloidosis. Interstitial depositions of amyloid are evident at Congo red staining, appearing as green birefringence under polarized light (×40)
Echocardiography represents a main diagnostic tool in CA. HF is frequently the result of the stiff-heart syndrome characterized by severe impairment of diastolic function secondary to amyloid infiltration of the myocardium, with relatively preserved systolic function and without cardiac remodelling [3, 4, 8–15]. However, it is important to realize that echocardiography cannot make the diagnosis in isolation, and images should be interpreted in the context of the clinical picture and other investigations. Moreover, the different types of amyloidosis that affect the heart (i.e., AL and variant/wild-type TTR types) cannot be distinguished by echocardiographic features, and this differential diagnosis is dependent on the recognition of some clinical and humoral clues and must be confirmed by immunohistochemical analysis from biopsy specimen [8]. Identifying the specific type of amyloidosis is compulsory in this setting, because therapeutic options are very different.
The most common echocardiographic feature is a concentric thickening of the left ventricular (LV) walls (Fig. 20.3, Clips 20.1 and 20.2), particularly in the absence of secondary causes. The combined presence of increased LV mass and low voltages at ECG (Fig. 20.1) may be more specific for infiltrative disease [4, 9, 16, 17]. Rahman et al. [9] studied 196 patients referred for EMB because of clinical suspicion of CA. The diagnosis was confirmed in 58 patients and, in multivariate logistic regression models, a combination of a low voltages and measures of myocardial thickness produced the most statistically useful model.
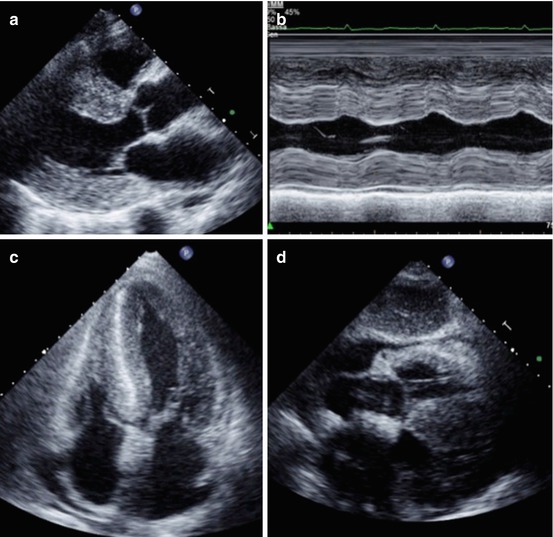
Fig. 20.3
Echocardiogram from a 72-year-old woman with cardiac amyloidosis. The left ventricle (LV) is severely hypertrophic (interventricular septum thickness 26 mm). Also, thickening of the right ventricle (RV) is evident. A granular sparkling appearance of the ventricular myocardium is evident. There is moderate thickening of valve leaflets and inter-atrial septum. Pericardial effusion is present. Parasternal long-axis view (a); M-mode echocardiogram of the left ventricle (b); apical four-chamber view (c); subcostal view (d)
LV systolic function is usually preserved until the late stages of the disease process. However, despite a normal LV ejection fraction (EF), longitudinal systolic function may be altered early during disease progression. In the presence of segmental wall-motion abnormalities (WMA) or global LV systolic dysfunction, a concomitant involvement of coronary arteries (due to atherosclerotic heart disease and/or coronary infiltration by amyloid) should be suspected [18].
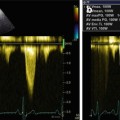
The echocardiographic appearance of CA can closely mimic several other diseases. Asymmetric hypertrophy of the septum due to amyloid deposition, even if rare, may occur, simulating HCM. Increased echogenicity of the myocardium (granular sparkling appearance) is not specific for CA and can also be found in myocarditis with severe fibrosis; it is quite common in HCM, as well as in other infiltrative and storage CMP [19]. Cardiac valves, papillary muscles, and intra-atrial septum are also frequently thickened, and pericardial effusion is frequently observed (Fig. 20.3, Clips 20.1 and 20.2). Mild valvular dysfunction is quite common, but severe dysfunction is rare [20]. Both atria are often enlarged, in keeping with diastolic dysfunction, which can involve both ventricles. The right ventricle (RV) can be involved in CA, frequently showing hypertrophy and dysfunction. Ghio et al. [21] studied 74 patients with AL amyloidosis and showed that 20 % of them had RV dysfunction [by tricuspid annular proximal systolic excursion (TAPSE)]. Diastolic dysfunction may be present in various degrees in CA. Doppler echocardiography has a main role in this setting. Advanced disease is frequently characterized by a restrictive filling pattern (RFP) (Fig. 20.4). In 1989, Klein et al. [22] were the first to describe the characteristics of LV filling pattern in patients with CA using Doppler echocardiography.
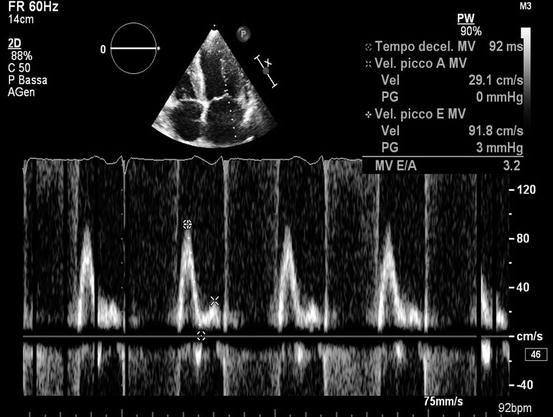
Fig. 20.4
Echocardiogram from a 50-year-old woman with cardiac amyloidosis. Transmitral pulsed Doppler shows severe diastolic dysfunction with restrictive filling pattern (E-wave velocity = 92 cm/s, A-wave velocity = 29 cm/s, E/A ratio = 3.2, E-wave deceleration time = 92 ms)
The above described echocardiographic abnormalities were reported and are seen in patients affected by symptomatic, clinically evident, CA and usually in an advanced stage. Efforts have been employed to identify cardiac involvement in the early stage of the disease in asymptomatic or mildly symptomatic patients.
Tissue-Doppler imaging (TDI) has shown reduced diastolic velocities in both early and late cardiac amyloid, so even early diastolic dysfunction could be identified, even in presence of minimal wall thickening [23]. As in healthy controls, in CA patients, there is a gradient of tissue velocities from the base to the apex, but peak systolic tissue velocities at the base and mid ventricle are significantly lower in CA patients with advanced disease and HF in comparison with early-stage asymptomatic patients [24].
Doppler myocardial imaging could have a role also in assessing RV dysfunction. Bellavia et al. [25] showed that E’ velocity (at the RV free wall) measured using TDI, as well as TAPSE, are often reduced in patients with AL amyloidosis and normal echocardiogram and are independent predictors of death.
Furthermore, as mentioned in Chap. 19, TDI can have a role in distinguishing restrictive physiology due to amyloid from constrictive pericarditis (CP). Sometimes the differential diagnosis is not easy, because the two diseases share some clinical and morphologic features. Peak early diastolic velocity from the lateral mitral annulus by TDI are usually markedly reduced in patients with CA compared with CP and normal individuals, which suggests that this measurement can provide a clinically valuable distinction between these two conditions [26].
Advanced echocardiographic techniques also focus on segmental deformation and its role in the differential diagnosis between CA and other diseases that may have a similar appearance at 2D echocardiography. Such diseases comprise HCM, Fabry disease, and hypertensive heart disease. Sun et al. [27] reported that global longitudinal strain detected by 2D speckle-tracking analysis was significantly lower in patients with CA compared with healthy controls but also compared with individuals with LV hypertrophy caused by HCM or hypertensive heart disease. Interestingly, CA is characterized by typical regional variations in longitudinal strain from base to apex. A relative apical-sparing pattern (Fig. 20.5, Clip 20.3a, 20.3b, and 20.3c) of longitudinal strain is an easily recognizable, accurate, and reproducible method of differentiating CA from other causes of LV hypertrophy [28].
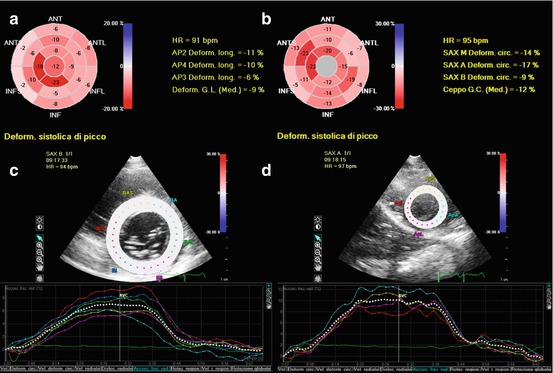
Fig. 20.5
Assessment of 2D speckle-tracking strain in a patient with amyloidosis. The left ventricle (LV) had a moderate hypertrophy (interventricular septum 14 mm, posterior wall 13 mm, mass 160 g/mq) and a moderate systolic dysfunction (ejection fraction 43 %). Strain imaging revealed global reduction of longitudinal (a), circumferential (b), and radial (c, d) strain, with a progressive increase in strain values from base to apex. Bull’s eye plot showing results of longitudinal strain analysis (a): basal LV level −5.3 %, mid level −7.1 %, apical level −14.5 %, and global −9 %. Bull’s eye plot with circumferential strain values (b): basal −9 %, mid −14 %, apical −17 %, and global −12 %. Radial strain assessed at basal LV (c) was 6.5 %, mid level 6.8 %, apical level 10 % (d), and global 7.8 %. ANT anterior, ANTL anterolateral, ANTS anteroseptal, AP2 apical two-chamber view, AP3 apical three-chamber view, AP4 apical four-chamber view, GC global circumferential (strain), GL global longitudinal (strain), HR heart rate, INF inferior, INFL inferolateral, INFS inferoseptal, SAX A short-axis apical view, SAX B short-axis basal view, SAX M short-axis mid view
Finally, Baccouche et al. [29] studied 12 patients with CA and 12 with HCM using 3D speckle-tracking echocardiography. These authors found important differences between the two diseases. In particular, the basal–apical radial strain gradient displayed oppositional characteristics in CA and HCM. The increasing basoapical radial strain gradient contrasts the physiological, decreasing basoapical radial strain gradient observed in healthy hearts [30].
CMR is particularly useful in CA imaging assessment by its value in morphological assessment, wall thickness evaluation, and the differential with other CMP [8, 31–34]. CMR morphofunctional study confirms the classic appearance of the disease, characterized by a restrictive configuration of the heart, with small and thick ventricles and large atria, frequent presence of pericardial and pleural effusions and/or ascites, and sometimes hypertrophy of the atrial septum [8, 33] (Fig. 20.6).
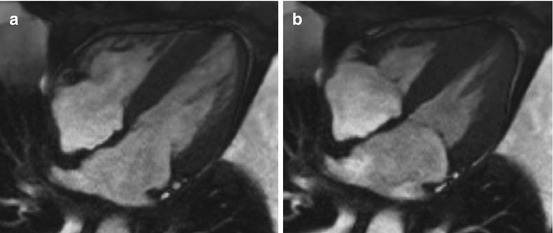
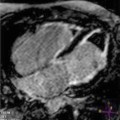
Fig. 20.6
Cardiac magnetic resonance (CMR) study in a 60-year-old patient affected by amyloid light-chain (AL) amyloidosis with cardiac involvement. End-diastolic (a) and end-systolic (b) frames. Note concentric LV thickening and severely impaired LV longitudinal function
In addition, CMR study can detect changes in myocardial tissue composition and architecture due to interstitial deposition of amyloid protein as changes in signal intensity on T1-weighted imaging. Furthermore, as myocardial intercellular amyloid deposition results in interstitial space expansion, late gadolinium-enhanced (LGE) CMR has main role in the diagnosis of CA. In fact, gadolinium chelates distribute in the extracellular space that is expanded by amyloid infiltration, leading to signal enhancement.
Syed et al. [34] performed LGE CMR in 120 patients referred to a tertiary center with histologically confirmed diagnosis of amyloidosis; 97 % of their CA patients had abnormal LGE, and 91 % had increased LV wall thickness on echocardiography. LGE pattern was global (transmural or subendocardial) in 83 % of cases (Fig. 20.7). Austin et al. [35] described a typical LGE pattern at CMR in CA patients, which is a diffuse circumferential LGE pattern involving the entire subendocardium and extending to adjacent myocardium. The diagnostic accuracy of this CMR pattern was superior to traditional noninvasive ECG and echocardiographic variables. However, LGE imaging has some limitations in CA, dealing in particular with contraindication in patients with severe renal impairment and by the fact that the above described patterns of LGE are often not uniform and are quite variable, leading to confusion in interpretation [34]. An interesting development in CMR is the advent of techniques that provide quantitative information on diffuse myocardial fibrosis by measuring the intrinsic magnetic resonance relaxation parameter T1 of the myocardium and mapping its spatial distribution. Karamitsos et al. [36] described a cohort of AL amyloidosis showing how native T1 correlates with systolic and diastolic dysfunction and has the potential to be more sensitive in detecting early disease than is LGE imaging.
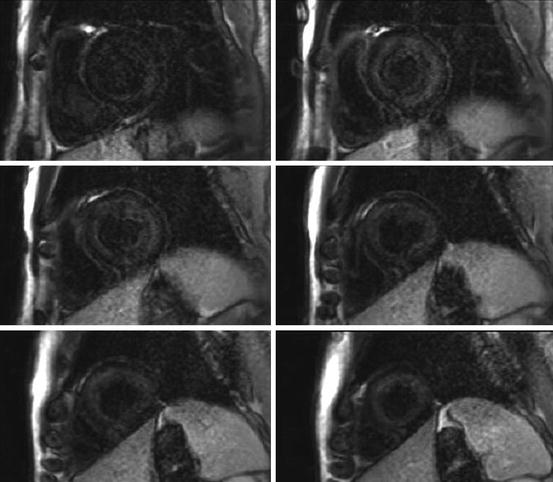
Fig. 20.7
Cardiac magnetic resonance (CMR) gadolinium-contrast-enhanced study in a patient with cardiac amyloidosis. The short-axis stack shows a global subendocardial late gadolinium enhancement (LGE) evident from the base to the apex of the left ventricle, corresponding to amyloid infiltration of myocardium
Another interesting new method that may play a future important role in assessing CA is extracellular volume measurement using CMR. Banypersad et al. [37] demonstrated the progressive increase of extracellular volume values from normals to patients with AL amyloidosis without cardiac involvement, and those with definite cardiac involvement. Thus, this measurement has potential to become the first noninvasive test able to quantify cardiac amyloid burden.
Amyloid deposits in the heart may be also detected by single-photon-emission computed tomography (SPECT) using technetium-99m (99mTc)-aprotinin, a protease inhibitor [38–40] and 99mTc-labelled phosphate derivatives, particularly in patients with hereditary transthyretin-related (TTR) amyloidosis [41–45]. The latter methods have important roles in differential diagnosis between TTR and AL amyloidoses and an improved capability in quantifying cardiac amyloid infiltration burden compared with LGE CMR [46].
An increased thallium-201 (201Tl)—a perfusion tracer—washout rate at rest–redistribution myocardial scintigraphy was reported in patients with CA and may reflect the severity of amyloid deposits in the myocardium [47]. SPECT studies with [123I]-metaiodobenzylguanidine (MIBG), an analog of norepinephrine, demonstrate myocardial dysinnervation (possibly due to the presence of amyloid deposits or a specific involvement of the autonomic system) both in patients with familial amyloidotic polyneuropathy [48, 49] and in those with AL amyloidosis and autonomic neuropathy [50]. Positron emission tomography (PET) with N-[methyl-11C]2-(4’-methylamino-phenyl)-6-hydroxybenzothiazole ([11C]-PIB), a tracer used to evaluate brain amyloidosis, was suggested as a method for studying systemic amyloidosis types AL and ATTR affecting the heart [51]. Parasympathetic myocardial denervation was also found in patients with familial amyloidotic polyneuropathy with [11C]-methylquinuclidinyl benzilate (MQNB) PET [49].
CA is historically considered a disease with poor short-term prognosis [3]. However, as demonstrated by Rapezzi et al. [8], it must not to be considered as a single entity, but the different underlying etiologies must be taken in account, especially when considering outcome, being that AL CA is the most aggressive form.
Echocardiography has a main role not only for determining cardiac involvement but in predicting outcome and monitoring cases over time. As shown by Klein et al. [22], Doppler-derived LV diastolic-filling variables and, in particular, the presence of RFP, are important predictors of survival in this disease. The same authors subsequently focused on the prognostic significance of a progression of LV-filling Doppler parameters during short-term follow-up that particularly affect patients with nonsevere thickening of LV walls [52]. Additional echocardiographic abnormalities, such as LA enlargement and RV dysfunction, were related with a worse prognosis in this disease [53]. Ghio et al. [21] found that RV dysfunction assessed by TAPSE was associated with a more severe LV involvement, higher plasma levels of N-terminal prohormone of brain natriuretic peptide (NT-proBNP) and poor prognosis. Speckle tracking showed that RV dysfunction seems to develop later than LV amyloid deposition, but, when it occurs, prognosis dramatically worsens [54]. In a large population of biopsy-proven AL CA patients, Buss et al. [55] showed that both TDI-derived longitudinal strain and speckle-tracking-derived global longitudinal strain were independent predictors of survival, with incremental power beyond standard clinical and serological parameters. The authors also demonstrated how NT-proBNP was strongly correlated with the parameters of longitudinal function considered above. Furthermore, Bellavia et al. [56], in a large cohort of patients with AL amyloidosis, identified peak longitudinal systolic strain of the basal anteroseptal segment as the main independent echocardiographic predictor of survival.
CMR, in addition to offering the clinician an excellent tool by which to attain a detailed view of morphological and functional abnormalities and to differentiate CA from other disease entities, may have a prognostic value [56]. First, this technique has an important role in early diagnosis of cardiac involvement, providing the opportunity to initiate early an adequate therapy [34, 57]. In addition, as reported by Austin et al. [35], the presence of typical diffuse circumferential LGE pattern at CMR is the only independent prognostic predictor on Cox proportional hazards analysis.
An additional clinical point must be considered in prognostic assessment of the disease. Although prognosis is generally poor in CA, long-term survival is rare but possible, and it is generally observed when the disease (and cardiac involvement) is detected at an early stage and adequate treatment is promptly implemented [58].
Finally, the underlying pathomechanisms of systolic and diastolic dysfunction associated with CA remain widely unknown. It has been hypothesized that a possible direct toxic effect of amyloid proteins could initially induce myocardial hypertrophy and then lead to systolic and/or diastolic dysfunction [59]. As the disease progresses, more amyloid deposits lead to atrophy and cardiomyocyte apoptosis and remodeling toward collagen formation and subsequent fibrosis [8]. Probably, this toxic effect is not equally present in all the types of amyloidosis. For example, Ng et al. [60] observed that patients with senile systemic amyloidosis (SSA) manifested a somewhat less aggressive disease course with respect to AL amyloidosis, despite the presence of more severe LV wall hypertrophy. A possible direct toxic effect related to the circulating monoclonal light chains in AL amyloidosis was hypothesized.
20.2.2 Sarcoidosis
Sarcoidosis is a chronic immune-mediated disease characterized by formation of granulomas in multiple organs (lungs, lymphatic system, liver, skin, eyes, nervous system, heart). Estimated prevalence is ~10.9 cases per 100,000 population [61]. The heart is involved in 20–30 % of cases, but only 5 % are clinically evident and symptomatic [62–64]. Every part of the heart can be involved in the disease. The predominant sites of myocardial involvement, in decreasing order of frequency, are LV free walls and papillary muscles, basal aspect of the interventricular septum (IVS), the RV wall, and atrial walls [63]. Clinical manifestations of cardiac sarcoidosis are variable and nonspecific; they range from asymptomatic conduction abnormalities to fatal ventricular arrhythmias, depending upon the location and extent of infiltration. Complete heart block is the most common finding (23–30 % of patients with clinically evident disease) [62–64]; first-degree heart block and bundle-branch blocks (BBB) also often occur due to involvement of the basal septum by scar tissue or granulomas or to that of the nodal artery, causing ischemia in the conduction system. Ventricular tachycardia is the most frequent arrhythmia in sarcoidosis (23 %), and sudden death (SD) secondary to ventricular arrhythmias has been described [63]; supraventricular arrhythmias are less common (15 % of cases). HF can be another manifestation and may be secondary to widespread infiltration of the myocardium, ventricular aneurysms, arrhythmias, cor pulmonale caused by pulmonary hypertension, valvular regurgitation, or a combination of these processes. End-stage HF is the cause of death in 25 % of patients affected by this disease, making it the second most frequent cause of death after SD (67 %). Other clinical manifestations include chest pain, pseudonecrosis Q waves at ECG, and pericardial abnormalities, such as pericardial effusion (3–19 % of cases), CP, and—rarely—cardiac tamponade [63].
The diagnosis of cardiac sarcoidosis can be very challenging from the clinical point of view. EMB, despite its specificity, has a low sensitivity as a result of patchy or focal infiltration [65]. The echocardiographic appearance of cardiac sarcoidosis varies among affected patients, ranging from normal to dilated ventricular chambers and normal to decreased regional and global systolic function. Ventricles may be globally hypokinetic, or the patchy nature of sarcoid infiltration of the heart may result in regional WMA [66]. Segmental WMA characteristically do not conform to any particular coronary distribution [66–68]. Two-dimensional echocardiographic morphologic abnormalities of cardiac sarcoid vary according to disease activity and include wall thickening due to granulomatous expansion and wall thinning due to fibrosis [66–68]. A typical site of these abnormalities, particularly wall thinning, is the basal anterior septum, the appearance of which in a young patient with dilated cardiomyopathy (DCM) is highly suggestive of sarcoidosis [67]. Scar retraction and aneurysms may develop, especially if the patient has been treated with corticosteroids. Sun et al. [69
 Dilated Cardiomyopathy: Usefulness of Imaging in Prognostic Stratification and Choice of Treatment
Dilated Cardiomyopathy: Usefulness of Imaging in Prognostic Stratification and Choice of Treatment
 Hypertrophic Cardiomyopathy: Clinical Assessment and Differential Diagnosis
Hypertrophic Cardiomyopathy: Clinical Assessment and Differential Diagnosis
 Definition, Classification, Epidemiology, and Clinical Relevance of Cardiomyopathies
Definition, Classification, Epidemiology, and Clinical Relevance of Cardiomyopathies
 Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Assessment and Differential Diagnosis
Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Assessment and Differential Diagnosis
 Basic Echocardiography in Hypertrophic Cardiomyopathy
Basic Echocardiography in Hypertrophic Cardiomyopathy
 Restrictive Cardiomyopathy: Clinical Assessment and Imaging in Diagnosis and Patient Management
Restrictive Cardiomyopathy: Clinical Assessment and Imaging in Diagnosis and Patient Management




Related posts:
Dilated Cardiomyopathy: Usefulness of Imaging in Prognostic Stratification and Choice of Treatment
Hypertrophic Cardiomyopathy: Clinical Assessment and Differential Diagnosis
Definition, Classification, Epidemiology, and Clinical Relevance of Cardiomyopathies
Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Assessment and Differential Diagnosis
Basic Echocardiography in Hypertrophic Cardiomyopathy
Restrictive Cardiomyopathy: Clinical Assessment and Imaging in Diagnosis and Patient Management
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
