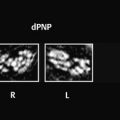
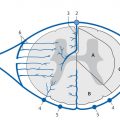
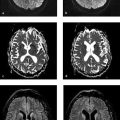
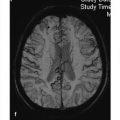
Neurodegenerative disease processes lead to the destruction of normally functioning neurons and their replacement by functionally deficient tissue such as glial scar tissue. In many cases the actual causes of neurodegenerative diseases are unknown. The classification of these diseases may be based on clinical, genetic, or pathologic-histologic criteria. For colleagues in clinical practice, especially neurologists, imaging studies are a key element in the diagnostic evaluation and follow-up of neurodegenerative diseases. The detection of typical brain changes may be crucial for directing further management, even though many of these disorders can only be treated symptomatically. This chapter is divided into three main parts based on clinical presentation. The first part deals with diseases of the central motor system, starting with the “archetypal” neurodegenerative process, Wallerian degeneration, in which a definite cause-and-effect relationship can be seen. It is followed by hypertrophic olivary degeneration, in which the cause-and-effect relationship is more complex but can still be understood in terms of neuroanatomic feedback loops. An actual cause cannot stated for many other of the diseases described. The second part of the chapter deals with Parkinson’s disease and its differential diagnosis. The third part explores various neurodegenerative forms of dementia. The individual diseases are defined or described in terms of their epidemiology, clinical presentation, treatment, and pathology. These points are followed by a description of their MRI findings and a list of differential diagnoses. Nuclear medicine studies such as positron emission tomography (PET) and single-photon-emission computed tomography (SPECT) make an important contribution to the differential diagnosis of some neurodegenerative diseases but are mentioned only briefly in this chapter. MRI is the most sensitive imaging modality for detecting neurodegenerative changes in the brain, which are often subtle. CT can add information by detecting calcifications. The degenerative process usually leads to scarring and atrophy. This is manifested by faint signal changes and a decrease in brain volume. None of the diseases described below are associated with abnormal enhancement that would indicate blood–brain barrier disruption. Thus, administration of intravenous contrast is useful only for the exclusion of other diseases. Methodology The following simple MRI protocol has proven effective in the diagnosis of neurodegenerative diseases: Axial T2w images. FLAIR images (e.g., sagittal). T1w images (e.g., coronal before and after intravenous contrast administration). Image planes tailored to the investigation (e.g., sagittal images for detecting pontine atrophy). Note With turbo spin echo (TSE) sequences, it is better to use a smaller turbo factor as this will optimize the echo time and ensure that even slight parenchymal signal changes can be detected. Optional sequences can be added, depending on the goal of the examination: T2*w or better susceptibility-weighted imaging (SWI); early signs for neurodegeneration. Very thin-slice T2w images (2–3 mm slice thickness, 512 matrix), e.g. to evaluate the brainstem or basal ganglia. Diffusion-weighted imaging (DWI), with calculation of the apparent diffusion coefficient (ADC). Magnetic resonance spectroscopy (MRS). Neuromelanin imaging (axial T1w images with thin slices). Definition Wallerian degeneration refers to the secondary antegrade degeneration of the axon and myelin sheath distal to the level at which the nerve cell has been severed from its nutrient perikaryon. Injured peripheral nerves may regenerate along their intact perineural sheath. In the central nervous system (CNS), however, complete regeneration cannot occur due to the formation of a glial scar, which blocks axonal growth. A typical cause of Wallerian degeneration in the CNS is an ischemic infarct involving the pyramidal tract. The degeneration is clearly visible, especially on coronal and sagittal images, owing to the close-packed arrangement of the parallel axons. MRI findings The causative parenchymal lesion is usually easy to identify. T2w hyperintensity is noted along the fiber tracts distal to the lesion, such as the pyramidal tract ( ▶ Fig. 8.1). This hyperintensity can be seen by 5 to 12 weeks after the causal event, and atrophy develops by 8 to 12 months. Fig. 8.1 Wallerian degeneration secondary to a lentiform nucleus infarction. (a) Axial T2w image (left) at the level of the basal ganglia demonstrates a fresh infarction of the right lentiform nucleus. The infarction appears hyperintense due to edema and has caused a slight mass effect. The posterior limb of the internal capsule is affected along with the right pyramidal tract. Another slice at the level of the cerebral peduncles (right) shows normal bilateral signal intensity of the pyramidal tract. (b) This series of axial T2w images, taken 1 year after the lentiform nucleus infarction in (a), documents the development of Wallerian degeneration along the right pyramidal tract, appearing as increased T2w signal intensity from the level of the infarction scar down into the decussation of the pyramidal tract. Differential diagnosis Tumors with extensive perifocal edema or microscopic infiltration of the surrounding brain parenchyma (e.g., by high-grade glioma) may spread from the supratentorial white matter along the pyramidal tract to the cerebral peduncle and brainstem. This process also appears hyperintense on T2w images. It is distinguished from Wallerian degeneration by noting edematous expansion of the pyramidal tract and the tumor mass effect on the supratentorial part of the pyramidal tract. This differs from the chronic mass lesion that results from degeneration. Note Following tumor surgery that has created large tissue defects bordering the corona radiata or pyramidal tract, it may be difficult to distinguish Wallerian degeneration from progressive microscopic tumor invasion. Definition A lesion in the myoclonic triangle ( ▶ Fig. 8.2; also know as the Guillain–Mollaret triangle) leads to secondary degeneration of the inferior olivary nucleus after a latent period of approximately 3 months (several weeks to several months). Frequent causes are an ischemic infarction or hemorrhage that involves the pontine tegmentum, with consequent damage to the central tegmental tract. A surgical cavity created by the excision of a brainstem cavernoma or pediatric cerebellar tumor, for example, may be the cause. Fig. 8.2 Myoclonic (Guillain–Mollaret) triangle. Schematic representation. The triangle consists of nerve pathways running from the dentate nucleus via the superior cerebellar peduncle in the dentatorubral tract to the contralateral red nucleus, then via the central tegmental tract to the ipsilateral olive. From there they run back to the dentate nucleus via the olivocerebral tract, inferior cerebellar peduncle, and contralateral cerebellar cortex. Clinical manifestations A palatal tremor, with rhythmic twitching of the soft palate at a frequency of 2 to 3 Hz, occurs on the side opposite to the affected olive. The tremor typically persists for years. Pathology Hypertrophy develops initially due to swelling of the olivary neurons and the formation of vacuoles. This is followed over a period of years by axonal demyelination, atrophy with neuronal loss, and glial scarring. MRI findings Images in the early stage show T2w hyperintensity in the anterior medulla oblongata at the level of the affected olive. Over the next few weeks, a volume increase is noted in that area due to swelling of the inferior olivary nucleus. The causative lesion in the triangle of Guillain and Mollaret, such as a tegmental infarct or small cerebellar hemorrhage, will appear as a scar due to the long latent period to onset of olivary degeneration ( ▶ Fig. 8.3). Hypertrophy of the olive may regress over a period of years, but the T2w hyperintensity will persist for life because of the scar tissue. Fig. 8.3 Hypertrophic olivary degeneration. (a) Axial T2w image (left) and coronal T2*w image (right) show remnants of a 5-month-old brainstem hemorrhage. The blood breakdown products appear in both images as susceptibility artifacts in the right half of the pontine tegmentum. Since the hemorrhage occurred, the patient has developed a palatal tremor on the left side. (b) Axial T2w image of the medulla oblongata. Secondary hypertrophic olivary degeneration appears as a hyperintense swelling on the right side. It results from damage to the right central tegmental tract at the level of the hemorrhage. Differential diagnosis Besides the symptomatic olivary degeneration described above, which may be unilateral or bilateral depending on lesion site, there is also an idiopathic form in which a causative lesion cannot be found. Moreover, studies have shown a possible association of hypertrophic olivary degeneration with other neurodegenerative diseases such as ▶ multisystem atrophy and progressive supranuclear palsy ( ▶ PSP). Epidemiology The prevalence of ALS is 5 to 10:100,000 population per year, with an approximately 3:1 male predominance. Over 90% of cases occur sporadically; autosomal dominant and recessive forms are rare. ALS may also be paraneoplastic, occurring most commonly in patients with bronchial, prostate, or breast cancer and in lymphoproliferative diseases. Clinical manifestations and treatment Usual age at onset is 40 to 60 years. Early signs are fasciculations in individual muscles, often including the tongue, accompanied by mixed flaccid and spastic paresis and muscular atrophy. Bulbar dysfunction is manifested by difficulties with speech and swallowing. Some patients experience compulsive laughter or crying. Approximately 80% of ALS patients die within 3 years, often by respiratory failure due to weakness of the respiratory muscles. Riluzole reportedly can prolong survival. Treatment is purely symptomatic and in the late stage may consist of long-term ventilation. Pathology ALS is caused by the degeneration of upper and lower motor neurons in the motor cortex, pyramidal tract, brainstem, and spinal cord. MRI findings Consistent with the underlying process of motor neuron degeneration with gliosis and demyelination, T2w images show hyperintensity along the pyramidal tract in the corona radiata, internal capsule, cerebral peduncles, and in the brainstem ( ▶ Fig. 8.4). Other possible findings are atrophy and reduced volume of the pyramidal tract and narrowing of the spinal cord. Asymmetrical atrophy of the tongue muscles may occur as an incidental finding. Fig. 8.4 ALS. Axial T2w images (a–d) from a patient in the early stage of ALS show faint, bilaterally symmetrical hyperintensity of the pyramidal tract in the corona radiata, internal capsule, and cerebral peduncle. The sagittal FLAIR image (e) also shows slight hyperintensity of the pyramidal tract in the corona radiata. (a) Axial T2w image. (b) Axial T2w image (next slice after a). (c) Axial T2w image (next slice after b). (d) Axial T2w image (next slice after c). (e) Sagittal FLAIR image. Differential diagnosis Clinical manifestation in the early stage of ALS are similar to those of spinal muscular atrophy, cervical myelopathy, and other motor system diseases. These conditions can be excluded by further testing. Often the subtle initial changes can be detected only by MRI follow-up. Epidemiology The prevalence of this disease, also known as Huntington’s chorea, is 4 to 7:100,000 population per year. It has an autosomal dominant mode of inheritance with full penetrance. The gene is located on the short arm of chromosome 4 and can be detected in trait carriers. Clinical manifestations and treatment The disease is manifested after age 40 and is often fatal within 10 to 20 years. Fully developed Huntington’s disease presents a triad of clinical symptoms: Choreiform movement disorders with sudden, jerking movements of the arms and legs, facial grimaces, and bizarre body postures. Behavioral changes with disinhibition and impulsive acts. Cognitive impairment progressing to dementia. There is no cure for Huntington’s disease, but treatment with neuroprotective agents may be tried. Pathology Atrophy with neuronal loss may be accompanied by astrocytosis. The atrophy is most pronounced in the caudate nucleus, putamen, and pallidum but may also affect the frontal lobes, hypothalamus, hippocampus, and the reticular part of the substantia nigra. MRI findings The relatively symmetrical atrophy of the striatum is best demonstrated in coronal images. Atrophy of the caudate nucleus causes its curvature to flatten while the lateral ventricles show corresponding enlargement ( ▶ Fig. 8.5). The striatum may be hyperintense in FLAIR and T2w images. Follow-ups will show progression of the findings over a period of years. Even in early cases, MRS will show a reduction of N-acetylaspartate in the basal ganglia. It may also show a reduction of creatine and a slight elevation of choline and possibly lactate. Fig. 8.5 Huntington’s disease. Coronal T1w images of a patient with Huntington’s disease (a) compared with images from a healthy same-age control subject (b). The Huntington images show frontal atrophy predominantly affecting the caudate nucleus. The coronal images show dilatation of the frontal horns of the lateral ventricles. Axial T2w images in Huntington’s disease show frontal atrophy predominantly affecting the caudate nucleus (c) compared with the healthy control (d). (a) Coronal T1w image images (patient with Huntington disease).(b) Coronal T1w images (healthy same-age control). (c) Axial T2w images (patient with Huntington’s disease). (d) Axial T2w images (healthy same-age control). Differential diagnosis Choreiform movement disorders may also result from inflammatory, infectious, vascular, hypoxic, and toxic lesions of the caudate nucleus and the striatum as a whole (e.g., after a lacunar infarct). The symptoms may even be unilateral (hemichorea), depending on the lesion ( ▶ Fig. 8.6). Chorea minor (Sydenham’s disease) typically affects school-age girls, occurring as a parainfectious disease with a good prognosis. Other diseases include chorea gravidarum and choreoacanthocytosis. A relatively symmetrical striatum atrophy may be detectable after hypoxic or inflammatory lesions but is not progressive. Fig. 8.6 Symptomatic form of hemichorea in a 66-year-old man with choreiform movement disorders on the left side due to nonketotic hyperglycemia. The images demonstrate a right-sided lesion of the basal ganglia, which show decreased volume and hyperintensity in T2w images. In the axial T2w and PDw images (a–c), the head of the caudate nucleus and putamen on the right side show decreased volume and high signal intensity. These structures show T1w hyperintensity in the coronal images (d), which is typical of nonketotic hyperglycemia. Petechial hemorrhages are the presumed cause. The symptoms and lesions usually resolve over a period of months. (a) Axial T2w image (magnified view of the midlevel slice in b).
8.2 Magnetic Resonance Imaging
8.3 Neurodegenerative Diseases of the Central Motor System
8.3.1 Wallerian Degeneration

8.3.2 Hypertrophic Olivary Degeneration


8.3.3 Amyotrophic Lateral Sclerosis

8.3.4 Huntington’s Disease



Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


