3. Recipients of contaminated blood products (3%)
4. Sexual partner of drug abuser + bisexual man
5. Infants born to a woman infected with HIV virus
◊ HIV antibodies present in > 50% of homosexuals + 90% of IV drug abusers!
◊ Rate of heterosexual transmission is increasing!
Clinical classification:
| group I | acute HIV infection with seroconversion |
| group II | asymptomatic HIV infection |
| group III | persistent generalized lymphadenopathy |
| group IV | other HIV disease |
| › | subgroup A | constitutional disease |
| › | subgroup B | neurologic disease |
| › | subgroup C | secondary infectious disease |
| › | subgroup D | secondary cancers |
| › | subgroup E | other conditions |
AIDS-defining pulmonary conditions (CDC, 1993):
(1) Tracheal / bronchial / pulmonary candidiasis
(2) Pulmonary CMV infection
(3) Herpes simplex bronchitis / pneumonitis
(4) Kaposi sarcoma
(5) Immunoblastic / Burkitt lymphoma
(6) Pneumocystis carinii pneumonia
(7) Mycobacterium tuberculosis / avium complex / kansasii
(8) Recurrent pneumonia
A. LYMPHADENOPATHY
Cause: reactive follicular hyperplasia = HIV adenopathy (50%), AIDS-related lymphoma (20%), mycobacterial infection (17%), Kaposi sarcoma (10%), metastatic tumor, opportunistic infection with multiple organisms, drug reaction
Location: mediastinum, axilla, retrocrural
B. OPPORTUNISTIC INFECTION
accounts for majority of pulmonary disease
◊ Pulmonary infection is often the first AIDS-defining illness!
1. Pneumocystis jirovecii pneumonia (60–80%) (formerly Pneumocystis carinii pneumonia)
20–40% develop > 1 episode during disease
• CD4+ T helper lymphocyte cell count ≤ 200/mm3
• subacute insidious onset with malaise, minimal cough
√ bilateral ground-glass infiltrates without effusion / adenopathy
√ bilateral perihilar interstitial infiltrates
√ diffuse bilateral alveolar infiltrates
√ frequently associated with pneumatoceles
√ apical predominance (in patients on prophylactic aerosolized pentamidine)
Mortality: in 25% fatal
2. Fungal disease (< 5%)
(a) Cryptococcus neoformans pneumonia (2–15%) usually associated with brain / meningeal disease
√ segmental infiltrate + superimposed pulmonary nodules ± lymphadenopathy ± pleural effusion
(b) Histoplasma capsulatum
√ typically diffuse nodular / miliary pattern at time of diagnosis
√ normal CXR in up to 35%
(c) Coccidioides immitis
√ diffuse infiltrates + thin-walled cavities
(d) Candida albicans
(e) Aspergillus: less common + less invasive ← relative preservation of neutrophilic function
› invasive pulmonary aspergillosis
› chronic necrotizing aspergillosis
› necrotizing tracheobronchitis
› obstructing bronchopulmonary aspergillosis
3. Mycobacterial infection (10% per year)
(a) M. tuberculosis (increasing frequency)
◊ AIDS patients are 500 times more likely to become infected than general population!
√ postprimary TB pattern with upper-lobe cavitating infiltrate
• CD4 lymphocyte count of 200–500 cells/mm3
√ primary TB pattern with lung infiltrate / lung masses + hilar / mediastinal lymphadenopathy + pleural effusion
• CD4 lymphocyte count of 50–200 cells/mm3)
√ atypical TB pattern with diffuse reticular / nodular infiltrates (CD4 lymphocyte count of < 50 cells/mm3)
√ adenopathy of low attenuation + rim enhancement
(b) M. avium-intracellulare (5%)
• in patients with low CD4 lymphocyte count only
√ diffuse bilateral reticulonodular infiltrates
√ adenopathy, miliary disease
(c) M. kansasii and others
4. Bacterial pneumonia (5–30%):
(a) Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus
√ frequently multilobar distribution
• bacteremia (common)
(b) Nocardia pneumonia (< 5%)
usually occurs in cavitating pneumonia
√ segmental / lobar alveolar infiltrate ± cavitation ± ipsilateral pleural effusion
(c) Rhodococcus equi (aerobic, Gram-negative)
√ cavitary pneumonia
(d) Bartonella henselae (Gram-negative)
= bacillary angiomatosis
• cutaneous lesions
√ highly vascular small pulmonary nodules
√ dramatic enhancement of enlarged lymph nodes
5. CMV pneumonia
most frequent infection found at autopsy (49–81%), diagnosed before death in only 13–24%;
high prevalence combined with Kaposi sarcoma
• fever, nonproductive cough, dyspnea, hypoxia
√ bilateral hazy infiltrates, focal nodules, masses
√ scattered widespread ground-glass opacities
√ thickening of bronchovascular bundles
√ tree-in-bud pattern
√ bronchiectasis / bronchial wall thickening
6. Toxoplasmosis
√ coarse interstitial / nodular pattern
√ focal areas of consolidation ± cavities
DDx: indistinguishable from PCP
C. TUMOR
1. Kaposi sarcoma (15%)
2. AIDS-related lymphoma of B-cell origin (2–5%) primarily immunoblastic NHL / Burkitt lymphoma / non-Burkitt lymphoma; occasionally Hodgkin disease
Location: pulmonary involvement (8–15%), CNS, GI tract, liver, spleen, bone marrow
Site: primarily extranodal
√ pleural effusion (50%)
√ hilar / mediastinal adenopathy (25%) ± axillary / supraclavicular / cervical adenopathy
√ solitary / multiple well-defined pulmonary nodules (occasionally with doubling times of 4–6 weeks)
√ diffuse bilateral reticulonodular heterogeneous opacities
√ alveolar infiltrates
√ paraspinal masses
D. LYMPHOID INTERSTITIAL PNEUMONITIS
Age: in children < 13 years of age
E. SEPTIC EMBOLI
F. PREMATURE DEVELOPMENT OF BULLAE (40%) with disposition to spontaneous pneumothorax
AIDS-related Complex (ARC)
= GENERALIZED LYMPHADENOPATHY SYNDROME
= prodromal phase of HIV seropositivity, generalized lymphadenopathy, CNS diseases other than those associated with AIDS
Time interval: ~ 10 years between seroconversion + clinical AIDS
• weight loss, malaise, diarrhea
• fever, night sweats, lymphadenopathy
• lymphopenia with selective decrease in helper T-cells
ADULT RESPIRATORY DISTRESS SYNDROME
= SHOCK LUNG = POSTTRAUMATIC PULMONARY INSUFFICIENCY = HEMORRHAGIC LUNG SYNDROME
= RESPIRATOR LUNG = STIFF LUNG SYNDROME = PUMP LUNG = CONGESTIVE ATELECTASIS = OXYGEN TOXICITY = ACUTE RESPIRATORY DISTRESS SYNDROME
= severe unexpected life-threatening acute respiratory distress characterized by abrupt onset of marked dyspnea, increased respiratory effort, severe hypoxemia associated with widespread airspace consolidation
Etiology:
◊ ARDS is the most severe form of permeability edema associated with diffuse alveolar damage
A. PRIMARY = DIRECT INJURY (← pulmonary disease)
= exposure to chemical agents, infectious pathogens, gastric fluid, toxic gas
Associated with: pulmonary consolidation
B. SYSTEMIC CONDITION (← extrapulmonary disease)
= sepsis, pancreatitis, severe trauma, blood transfusion → systemic biochemical cascade creating oxidating agents, inflammatory mediators, enzymes
Associated with: interstitial edema, alveolar collapse
Histo:
| › | up to 12 hr: | fibrin + platelet microemboli |
| › | 12–24 hr: | interstitial edema |
| › | 24–48 hr: | capillary congestion, extensive interstitial + alveolar proteinaceous edema + hemorrhage, widespread microatelectasis, destruction of type I alveolar epithelial cells |
| › | 5–7 d: | extensive hyaline membrane formation, hypertrophy + hyperplasia of type II alveolar lining cells |
| › | 7–14 d: | extensive fibroblastic proliferation in interstitium + within alveoli, rapidly progressing collagen deposition + fibrosis; almost invariably associated with infection |
Predisposed:
hemorrhagic / septic shock, massive trauma (pulmonary / general body), acute pancreatitis, aspiration of liquid gastric contents, heroine / methadone intoxication, massive viral pneumonia, traumatic fat embolism, near-drowning, conditions leading to pulmonary edema
mnemonic: DICTIONARIES
Disseminated intravascular coagulation
Infection
Trauma
Inhalants: smoke, phosgene, NO2
O2 toxicity
Narcotics + other drugs
Aspiration
Radiation
Includes pancreatitis
Emboli: amniotic fluid, fat
Shock: septic, hemorrhagic, cardiogenic, anaphylactic
• initially few / no symptoms
• rapidly progressive dyspnea, tachypnea, cyanosis
• hypoxia unresponsive to oxygen therapy ← AV shunting
• NO increase in pulmonary capillary pressure
Stages (often overlapping):
1st (early / exudative) stage
= interstitial edema with high protein content rapidly filling the alveolar spaces associated with hemorrhage → ensuing hyaline membrane formation
√ interstitial edema (with high protein content) initially
√ perihilar areas of increased opacity following rapidly
√ widespread alveolar consolidation with predominantly peripheral cortical distribution
√ air bronchogram
√ gravitational gradient (best seen on CT) ← dependent atelectasis
DDx: cardiogenic edema (cardiomegaly, apical vascular distribution, Kerley lines)
2nd (proliferative) stage
= organization of fibrinous exudate + subsequent regeneration of alveolar lining + thickening of alveolar septa
√ inhomogeneous areas of patchy / diffuse ground-glass opacities
3rd (fibrotic) stage (after days to weeks)
= varying degrees of scarring
√ dependent gradient of consolidation (often)
√ bronchial dilatation with improvement / even resolution
√ ± progression to irreversible varicoid bronchial enlargement
√ subpleural and intrapulmonary cysts
Cx: pneumothorax
CXR:
√ NO cardiomegaly / pleural effusion
› up to 12 hours:
√ characteristic 12-hour delay between clinical onset of respiratory failure and CXR abnormalities
› 12–24 hours:
√ patchy ill-defined opacities throughout both lungs
› 24–48 hours:
√ massive airspace consolidation of both lungs
› 5–7 days:
√ consolidation becomes inhomogeneous ← resolution of alveolar edema
√ local areas of consolidation ← pneumonia
› > 7 days:
√ reticular / bubbly lung pattern ← diffuse interstitial + airspace fibrosis
ALPHA-1 ANTITRYPSIN DEFICIENCY
= rare autosomal recessive disorder
Source: alpha-1 antitrypsin (glycoprotein) is to > 90% synthesized in hepatocytes + released into serum
Gene: codominant gene expression on chromosome 14 with > 100 genetic variants of the protein; most severe hepatopulmonary manifestations result from homozygous PiZZ phenotype
Action: proteolytic inhibitor of neutrophil elastase, trypsin, chymotrypsin, plasmin, thrombin, kallikrein, leukocytic + bacterial proteases; neutralizes circulating proteolytic enzymes
Mode of injury from deficiency:
PMNs + alveolar macrophages sequester into lung during recurrent bacterial infections + release neutrophil elastase, which acts unopposed + digests basement membrane
Age: early age of onset (20–30 years); M÷F = 1÷1
• rapid + progressive deterioration of lung function:
• dyspnea in 4th and 5th decade
◊ 20% of homozygotic individuals never develop clinically apparent emphysema
• chronic sputum production (50%)
√ severe panacinar emphysema with basilar predominance ← gravitational distribution of pulmonary blood flow:
√ ↓ in size + ↓ in number of pulmonary vessels in lower lobes
√ redistribution of blood flow to unaffected upper lung zones
√ bullae at both lung bases
√ marked flattening of diaphragm
√ minimal diaphragmatic excursion
√ multilobar cystic bronchiectasis (40%)
√ hepatopulmonary syndrome
Cx: hepatic cirrhosis (in homozygotic individuals)
◊ Most common metabolic liver disease in children
Prognosis: 15–20-year decrease in longevity in smokers relative to nonsmokers
ALVEOLAR MICROLITHIASIS
= very rare disease of unknown etiology characterized by myriad of calcospherites (= microliths composed of calcium + phosphorus) within alveoli
Cause: autosomal recessive inheritance of a gene mutation preventing production of a transporter protein for sodium-dependent transfer of phosphate into type II pneumocytes → phosphorus ions stay in alveolar space
Age peak: 30–50 years; begins in early life; has been identified in utero; M÷F = 1÷1; in 50% familial (in at least one sibling)
• usually asymptomatic (70%)
• progressive dyspnea on exertion, cyanosis, clubbing of fingers
• striking discrepancy between marked radiographic findings and mild clinical symptoms
• NORMAL serum calcium + phosphorus levels
• restrictive lung disease (↓ in residual volume and ↓ diffusion capacity)
Distribution: predisposition for posterior segments of lower lobes + anterior segments of upper lobes; medial lung >> lateral lung
√ very fine, sharply defined, sandstorm-like micronodular (< 1 mm) pattern
√ diffuse involvement of both lungs
√ obscuration of mediastinal + diaphragmatic reflections
√ vertical linear radiolucency between ribs + lung parenchyma ← subpleural cystic changes
√ intense uptake on bone scan
HRCT:
√ thickening + micronodulation of interlobular septa ← innumerable microliths within periphery of secondary pulmonary lobule
√ thickened micronodular appearance of bronchovascular + subpleural interstitium
√ pleural calcifications
√ ground-glass appearance (of microliths < 1 mm) ± “crazy paving” pattern
(a) late development of pulmonary insufficiency ← interstitial fibrosis + cor pulmonale
(b) disease may become arrested
(c) microliths may continue to form / enlarge
Rx: NO effective treatment aside from lung transplant
Dx: open lung biopsy
DDx: (1) Pulmonary alveolar proteinosis, sarcoidosis, pneumoconiosis, pulmonary hemosiderosis, amyloidosis, miliary tuberculosis, metastatic pulmonary calcifications of renal failure
(2) “Mainline” pulmonary granulomatosis (IV abuse of talc-containing drugs such as methadone, rarely as numerous + scarring + loss of volume)
ALVEOLAR PROTEINOSIS
= PULMONARY ALVEOLAR PROTEINOSIS (PAP)
= rare disorder characterized by accumulation of lipoproteinaceous material in alveoli ← altered surfactant homeostasis
Forms:
A. CONGENITAL PAP
B. PRIMARY ACQUIRED PAP (90%)
Age: median 39 years; M>> F
Predisposed: smoking in 72%
C. SECONDARY PAP
Cause: hematologic cancers, pharmacologic immunosuppression, inhalation of inorganic dust (eg, silica), toxic fumes, certain infections
Pathophysiology: functional impairment / reduced number of alveolar macrophages
Etiology: ?; associated with dust exposure (eg, silicoproteinosis is histologically identical to PAP), immunodeficiency, hematologic + lymphatic malignancies, AIDS, chemotherapy
Pathophysiology:
(a) overproduction of surfactant by granular pneumocytes
(b) defective clearance of surfactant by alveolar macrophages
Histo: alveoli filled with proteinaceous material (the ONLY pure airspace disease), normal interstitium
Age peak: 39 years (range, 2–70 years); M÷F = 3÷1
• asymptomatic (10–20%), gradual onset of dyspnea + cough
• weight loss, weakness, hemoptysis, defect in diffusing capacity
√ bilateral air-space disease with ill-defined nodular / confluent ground-glass pattern:
√ perihilar predominance of “bat-wing” configuration WITHOUT signs of left-sided heart failure
√ small acinar nodules + coalescence + consolidation
√ patchy peripheral / primarily unilateral infiltrates (rare)
√ reticular / reticulonodular / linear interstitial pattern with Kerley B lines (late stage)
√ slow clearing over weeks or months
√ slow progression (⅓), remaining stable (⅔)
√ NO adenopathy, NO cardiomegaly, NO pleural effusion
HRCT:
√ crazy-paving pattern = combination of patchy ground-glass opacities + smooth interlobular septal thickening often in geographic distribution
√ sharp demarcation between normal + abnormal lung
√ consolidation
Cx: susceptible to pulmonary infections ← poorly functioning macrophages + excellent culture medium for opportunistic pathogens (esp. Nocardia asteroides), other common respiratory pathogens
Prognosis:
highly variable course with clinical and radiologic episodes of exacerbation + remissions
(a) 50% improvement / recovery
(b) 30% death within several years under progression
Dx: bronchoalveolar lavage, transbronchial / open lung biopsy
Rx: bronchopulmonary lavage
DDx:
(a) during acute phase: pulmonary edema, diffuse pneumonia, ARDS
(b) in chronic stage:
1. Idiopathic pulmonary hemosiderosis (boys, symmetric involvement of mid + lower zones, progression to nodular + linear pattern)
2. Hemosiderosis (bleeding diathesis)
3. Pneumoconiosis
4. Hypersensitivity pneumonitis
5. Goodpasture syndrome (more rapid changes, renal disease)
6. Desquamative interstitial pneumonia (“ground-glass” appearance, primarily basilar + peripheral)
7. Pulmonary alveolar microlithiasis (widespread discrete intraalveolar calcifications primarily in lung bases, rare familial disease)
8. Sarcoidosis (usually with lymphadenopathy)
9. Lymphoma
10. Bronchioloalveolar cell carcinoma (more focal, slowly enlarging with time)
AMNIOTIC FLUID EMBOLISM
= most common cause of maternal peripartum death
• dyspnea, shock during / after labor + delivery
Pathogenesis: amniotic debris enters maternal circulation resulting in:
(1) Pulmonary embolization
(2) Anaphylactoid reaction
(3) DIC
√ usually fatal before radiographs obtained
√ may demonstrate pulmonary edema
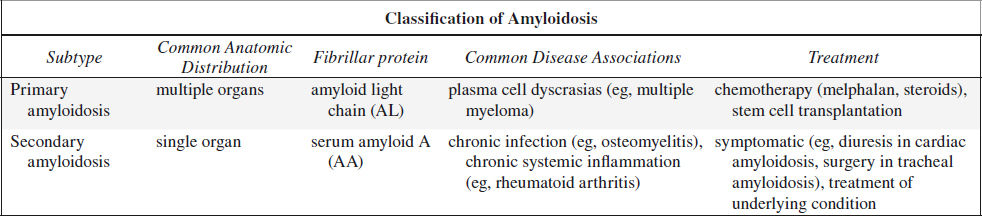
AMYLOIDOSIS OF THE CHEST
= disease characterized by an extracellular deposition of insoluble fibrillar proteins aggregating into twisted ß-pleated sheets of great chemical diversity
Histo: protein (immunoglobulin) / polysaccharide complex; affinity for Congo red stain
Pulmonary Amyloidosis
Frequency: 1° amyloidosis (in up to 70%), 2° amyloidosis (rare)
A. NODULAR PARENCHYMAL TYPE (most common)
Age: 50–60 years of age; M÷F = 1÷1
• usually asymptomatic, incidental discovery on CXR
Size: 5–15 cm
Site: subpleural / peripheral
Distribution: concentrated in lower lobes
√ solitary mass = amyloidoma (60%)
√ multiple pulmonary nodules:
√ smooth lobulated / spiculated margins
√ cavitation (rare)
√ ± central calcification / ossification (in up to 50%)
DDx: chronic renal failure
√ mediastinal / hilar adenopathy
Dx: AL amyloid (= amyloid light chain) on biopsy
Prognosis: excellent with slow progression over years; incurable; treatment rarely required
Rx: low-dose prednisone, colchicine
DDx: metastatic disease, granulomatous disease (fungal, TB), rheumatoid lung, sarcoidosis, mucoid impaction, bronchogenic carcinoma, chondroma
B. DIFFUSE ALVEOLAR SEPTAL TYPE (least common)
◊ Most commonly associated with systemic amyloidosis
Age: > 60 years of age
May be associated with: concurrent systemic involvement
• usually asymptomatic with normal CXR
• cough + dyspnea with abnormal CXR
CT:
√ well-defined scattered 2–4 mm micronodules
√ reticulations = widespread small irregular densities (exclusively interstitial involvement) ± calcification
√ interlobular septal thickening
√ may become confluent ± honeycombing
Distribution: basal + peripheral predominance
√ punctate lung calcifications
√ pleural thickening
√ ± pleural effusion
√ rarely associated with thin-walled cysts (most commonly in Sjögren syndrome)
Dx: AL amyloid (= amyloid light chain) on biopsy
Prognosis: median survival of 16 months ← pulmonary hypertension, respiratory failure
DDx: pneumoconiosis (especially asbestosis), idiopathic pulmonary fibrosis, rheumatoid lung, Langerhans cell histiocytosis, scleroderma, cancer with lymphangitic spread
Airway Amyloidosis (2nd most common manifestation)
= TRACHEOBRONCHIAL TYPE
= diffuse / solitary (rarely) submucosal deposition of amyloid in trachea + segmental airways
Age: 50–70 years; M > F
Rarely associated with: systemic amyloidosis
• hemoptysis (most frequent complaint)
• stridor, cough, dyspnea, hoarseness, wheezing
CXR: notoriously insensitive
CT:
√ diffuse rigid narrowing of a long tracheal segment (best depicted on COR and SAG reconstructions)
√ calcified / ossified tracheal wall also affecting posterior membrane of trachea (DDx to tracheobronchopathia osteochondroplastica / relapsing polychondritis)
√ luminal narrowing by multiple nodules protruding from wall of trachea / large bronchi
√ prominent bronchovascular markings
√ obstruction with consolidation
√ atelectasis
√ hyperinflation
√ bronchiectasis
√ destructive pneumonitis
PET/CT:
√ increased FDG uptake may allow detection of early amyloidosis + response to treatment
Cx: recurrent pneumonia
Rx: local bronchoscopic resection, laser therapy, stent placement, radiation therapy
Prognosis: 30–50% 5-year survival; worse for proximal disease
DDx: endobronchial neoplasm
Mediastinal Amyloidosis (3rd most common manifestation)
Commonly associated with: systemic amyloidosis
Location: multistation lymphadenopathy / anatomically localized isolated tumefactive amyloidoma
• asymptomatic lymphadenopathy (frequent)
√ enhancing lymphadenopathy
√ punctate / diffuse / eggshell lymph node calcifications
ASBESTOS-RELATED DISEASE
Substances:
Aspect (length-to-diameter) ratio effects carcinogenicity:
eg, aspect ratio of 32 = 8 µm long x 0.25 µm wide
› commercial amphiboles: straight, rigid, needlelike
• crocidolite = blue / black asbestos
• amosite = brown asbestos
› commercial serpentines (= nonamphiboles):
• chrysotile = white asbestos (the only mineral in the serpentine group accounting for > 90% of asbestos used in the USA)
› noncommercial contaminated amphiboles
• actinolite; anthophyllite; tremolite
(a) relatively benign:
(1) Chrysotile in Canada
(2) Anthophyllite in Finland, North America
(3) Tremolite
(b) relatively malignant:
(1) Crocidolite in South Africa, Australia
(2) Amosite
◊ Very fine fibers (crocidolite) are associated with the largest number of pleural disease!
◊ Asbestos fibers up to 100 µm in length
Occupational exposure:
(a) asbestos mining, milling, processing
(b) insulation manufacturing, textile manufacturing, construction and demolition, pipe fitting, shipbuilding, gaskets, brake linings
Pathophysiology:
asbestos-activated macrophages produce a variety of growth factors that interact to induce fibroblast proliferation; oxygen-free radicals released by macrophages damage proteins + lipid membranes sustaining the inflammatory process
Asbestos-related Pleural Disease
1. Pleural Effusion (21%)
= earliest asbestos-related pleural abnormality, frequently followed by diffuse pleural thickening + rounded atelectasis
Prevalence: 3% (higher with increasing levels of asbestos exposure)
Latency period: 8–10 years after exposure
• benign asbestos pleurisy:
• may be associated with chest pain (⅓)
• usually small sterile serous / hemorrhagic exudate
√ recurrent bilateral effusions
√ ± plaque formation
DDx: TB, mesothelioma
2. Focal Pleural Plaques (65%)
= hyalinized collagen in submesothelial layer of parietal pleura
◊ Most useful marker of asbestos exposure!
Frequency: most common manifestation of exposure; 6% of general population will show plaques; 3–14% of dockyard workers; 58% of insulation workers
Latency period: in 10% (50%) after 20 (40) years
Histo: dense hypocellular undulating collagen fibers often arranged in a “basket weave” pattern ± focal / massive calcifications; may contain large numbers of asbestos fibers (almost exclusively chrysotile)
Location: bilateral + multifocal; posterolateral chest wall between 7th–10th rib following rib contours; lateral chest wall between 6th–9th rib; aponeurotic dome of diaphragm; mediastinum
◊ Apices + costophrenic angles are spared!
◊ RARE in fissures of lung (visceral pleura)
Site: parietal pleura (visceral pleura typically spared)
• asymptomatic, no functional impairment
CXR:
√ geographic / holly leaf-shaped veil-like opacity
√ localized sharply marginated pleural thickening
CT (more sensitive + specific):
√ usually focal nodular area of pleural thickening (< 1 cm thick) with edges thicker than central portions of plaque; in 48% only finding; in 41% with parenchymal changes; stable over time
√ thin layer of extrapleural fat separates plaque from underlying rib + intercostal muscle
√ “hairy plaque” = visceral pleural plaque + underlying short interstitial lines radiating from plaque (rare)
√ no hilar adenopathy
√ calcified (in 10–15% by X-ray, in 15–20% by CT)
Prognosis: NO risk of malignant degeneration; increased risk of developing mesothelioma + bronchogenic carcinoma
DDx: chest wall fat, rib fractures, rib companion shadow
3. Pleural Calcification (21–25–60%)
◊ HALLMARK of asbestos exposure!
◊ detected by CXR (CT) in 25% (60%)
Latency: 20% (40%) become visible in > 20 (40) years
Histo: calcification starts in parietal pleura; calcium deposits may form within center of plaques
√ dense lines paralleling the chest wall, mediastinum, pericardium, diaphragm
◊ Bilateral diaphragmatic calcifications with clear costophrenic angles are PATHOGNOMONIC!
√ advanced calcifications are leaflike with thick-rolled edges
DDx: talc exposure, hemothorax, empyema, therapeutic pneumothorax for TB (often unilateral, extensive sheetlike, on visceral pleura)
4. Diffuse Pleural Thickening (17%)
= smooth uninterrupted diffuse thickening of parietal pleura extending over at least ¼ of chest wall (visceral pleura involved in 90%, but difficult to demonstrate)
• may cause restriction of pulmonary function
May be associated with: rounded atelectasis
√ bilateral process with “shaggy heart” appearance (20%)
√ smooth (difficult to assess when viewed en face)
√ thickening of interlobar fissures
√ focally thickened diaphragm
√ obliterated costophrenic angles (minority of cases)
DDx: pleural thickening from parapneumonic effusion, hemothorax, connective tissue disease
Pulmonary Asbestosis
= (term “asbestosis” reserved for) chronic progressive diffuse interstitial pulmonary fibrosis ← inhalation of asbestos fibers
Frequency: in 49–52% of industrial asbestos exposure; 1,229 new cases annually in USA (2013)
Latency period: 40–45 years; dose-effect relationship
Histo: interstitial fibrosis begins around respiratory bronchioles, then progresses to involve adjacent alveoli
Diagnostic criteria:
1. Reliable history of exposure
2. Appropriate time interval between exposure + detection
3. Radiographic opacities classified as ILO s,t,u
4. Restrictive pattern of lung impairment
5. Diffusing capacity below normal range
6. Bilateral crackles at posterior lung bases, NOT cleared by cough
• dyspnea
• restrictive pulmonary function tests: progressive reduction of vital capacity + diffusing capacity
• asbestos bodies in macrophages from bronchoalveolar lavage (BAL) fluid (= single asbestos fiber surrounded by segmented protein-iron coat)
Location: lower posterior bases > apices
Site: most severe in subpleural zones (asbestos fibers concentrate beneath visceral pleura)
CXR:
√ small irregular linear opacities (NOT rounded as in coal / silica) progressing from fine to coarse reticulations:
√ confined to lung bases, progressing superiorly
√ septal lines (= fibrous thickening around 2ndary lobules)
√ honeycombing (uncommon)
√ “shaggy” (obscured) heart border ← parenchymal + pleural changes
√ ill-defined outline of diaphragm
√ rarely massive fibrosis, predominantly at lung bases without migration toward hilum (DDx from silicosis / CWP)
√ ABSENCE of hilar / mediastinal adenopathy (if present consider other diagnosis)
HRCT:
◊ Obtain scan in prone position to differentiate from gravity-related physiologic phenomena
√ thickened intralobular lines as initial finding ← centrilobular peribronchiolar fibrosis:
√ multiple subpleural curvilinear branching lines (“subpleural pulmonary arcades”) = dotlike reticulonodularities connected to the most peripheral branch of pulmonary artery
Site: most prominent posteriorly parallel to and within 1 cm of pleura
√ thickened interlobular septal lines (= interlobular fibrotic / edematous thickening):
√ reticulations = network of linear densities, usually posteriorly at lung bases
√ architectural distortion of lobule
√ parenchymal band formation = linear < 5 cm long + several mm wide opacity, often extending to pleura, which may be thickened + retracted at site of contact
√ subpleural curvilinear lines
√ patchy areas of ground-glass attenuation (= alveolar wall thickening due to fibrosis / edema)
√ honeycombing = multiple cystic spaces < 1 cm in diameter with thickened walls
NUC:
√ 67Ga uptake gives a quantitative index of inflammatory activity
Cx: pulmonary fibrosis, pleuropulmonary malignancy (latency period of > 20 years)
DDx: idiopathic pulmonary fibrosis (NO parietal pleural thickening)
Atelectatic Asbestos Pseudotumor
= ROUND ATELECTASIS = “FOLDED LUNG”
= infolding of redundant pleura accompanied by segmental / subsegmental atelectasis
◊ Most common of benign masses caused by asbestos exposure!
Location: posteromedial / posterolateral basal region of lower lobes (most common); frequently bilateral
√ 2.5–8 cm focal subpleural mass abutting a region of thickened pleura
√ size + shape show little progression, occasionally ↓ in size
CT:
√ rounded / lentiform / wedge-shaped peripheral mass
√ pleural thickening ± calcification always present and frequently greatest near mass
√ “crow’s feet” = linear bands radiating from mass into lung parenchyma (54%)
√ “vacuum cleaner” / “comet” sign
= bronchovascular markings emanating from nodular subpleural mass + coursing toward ipsilateral hilum
√ “Swiss cheese” air bronchogram (18%)
√ partial interposition of lung between pleura + mass
√ volume loss of affected lobe ± hyperlucency of adjacent lung
Asbestos-related Malignancy
Estimated yearly asbestos-related deaths in USA (2013):
12,000–15,000 annually
Lung Cancer
Incidence: 180,000 new cases annually in USA; 20–25% of workers heavily exposed to asbestos
Occurrence related to:
(a) cumulated dose of asbestos fibers
(b) smoking (synergistic carcinogenic effect)
◊ 100-fold increased risk in smokers versus a 7-fold increased risk in nonsmokers!
(c) preexisting interstitial disease
(d) occupational exposure to known carcinogen
Latency period: 25–35 years
Associated with: increased incidence of gastric carcinoma
Histo: bronchioloalveolar cell carcinoma (most common); bronchogenic carcinoma (adenocarcinoma + SCC)
Location: at lung base / in any location if associated with smoking
Malignant Mesothelioma
Incidence: 2,686 new cases annually in USA (2013); 7,000-fold increase in incidence
Risk: 10% over lifetime of an asbestos worker; household members of asbestos worker; residents near asbestos mines and plants
Latency period: 20–40 years
Gastrointestinal Neoplasm
= pancreatic, liver, gallbladder, colon, rectal, stomach cancer
Frequency: 3-fold increase (weak link)
ASKIN TUMOR
= EXTRASKELETAL EWING SARCOMA = PRIMITIVE NEUROECTODERMAL TUMOR (PNET)
= uncommon tumor probably arising from intercostal nerves
Mean age: 14.5 years; M÷F = 1÷3; Caucasian
Path: neuroectodermal small cell tumor containing neuron-specific enolase (may also be found in neuroblastoma)
• chest wall mass with / without pain
• constitutional symptoms: fever, anorexia, weight loss
√ large tumor involving chest wall + pleura
√ high tumor vascularity
√ rib destruction (occasionally arising from rib) in 25–63%
√ often large malignant pleural effusion
√ pulmonary parenchymal disease (25%)
√ calcifications (10%)
√ ipsilateral hilar + mediastinal lymphadenopathy
√ ± pneumothorax ← pulmonary involvement
CT:
√ large unilateral heterogeneous chest wall mass with intra- and extrathoracic components
√ ± pleural, pericardial, diaphragmatic, vertebral, spinal extension and involvement
MR:
√ predominantly intermediate SI on T1WI + high SI on T2WI
√ prominent areas of high SI on T2WI ← hemorrhage + necrosis
√ commonly invasion of chest wall musculature + mediastinum + lung
NUC:
√ ↑ activity on bone scan + 111In-pentetreotide + 99mTc-MIBI
√ NO uptake on FDG PET
Metastatic to: lung, mediastinal nodes, bone (25%), CNS, liver, adrenal
◊ At presentation pulmonary metastases (in 38%) + mediastinal lymphadenopathy (in 25%)
Prognosis: median survival of 8 months
DDx: Ewing sarcoma, lymphoma, chest wall hamartoma in infancy
ASPERGILLOSIS
Organism:
Aspergillus fumigatus = intensely antigenic ubiquitous fungus in soil, water, decaying vegetable and animal matter existing as
(a) conidiophores = reproductive form releasing thousands of spores
(b) hyphae (= matured spores) characterized by 45° dichotomous branching pattern
Occurrence:
commonly in sputum of normal persons, ability to invade arteries + veins facilitating hematogenous dissemination
M÷F = 3÷1
Transmission: spore inhalation
Predisposed:
(a) preexisting lung disease (tuberculosis, bronchiectasis)
(b) impairment of immune system (alcoholism, advanced age, malnutrition, concurrent malignancy, poorly controlled diabetes, cirrhosis, sepsis)
• tracheo-bronchitis, bronchiolitis, bronchopneumonia
• fungal hyphae in lumen of airways
• positive precipitin test to Aspergillus antigen
• elevated Aspergillus-specific IgE, IgG-ELISA, polymerase chain reaction identification
Cx: dissemination to heart, brain, kidney, GI tract, liver, thyroid, spleen
◊ Sputum cultures are diagnostically unreliable because of normal (saprophytic) colonization of upper airways!
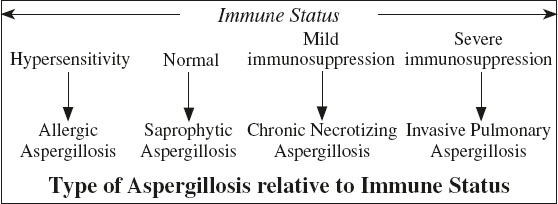
Noninvasive Aspergillosis = Mycetoma Formation
= SAPROPHYTIC ASPERGILLOSIS
= noninvasive colonization of preexisting cavity / cyst in immunologically normal patients with cystic / cavitary lung disease like:
granulomatous disease [sarcoidosis (common), remote TB, atypical mycobacterial infection, Pneumocystis jirovecii], bronchiectasis, cystic fibrosis, abscess, bullous emphysema, carcinoma, traumatic pneumatocele, ankylosing spondylitis, Marfan syndrome, neurofibromatosis type 1
• blood-streaked sputum / severe hemoptysis (45–95%)
• elevated serum precipitins level for Aspergillus (50%)
√ solid round gravity-dependent mass within preexisting spherical / ovoid thin-walled cavity (= Monad sign):
[monas, Greek = single, alone; – ad, Greek suffix = group, unit]
Histo: mycetoma = aspergilloma = fungus ball = masslike collection of intertwined hyphae matted together with fibrin, mucus, cellular debris colonizing a pulmonary cavity
√ “air-crescent” sign = crescent-shaped air space separating fungus ball from nondependent cavity wall
√ fungus ball may calcify in scattered / rimlike fashion
√ pleural thickening adjacent to preexisting cyst / cavity = commonly first sign before visualizing mycetoma
Cx: life-threatening hemoptysis with mycetoma
N.B.: search for hypertrophied bronchial arteries as road map for bronchial artery embolization
Dx: transthoracic needle biopsy / bronchial washings
DDx of other organisms causing fungus ball:
Candida albicans, Pseudallescheria boydii, Coccidioides immitis, Nocardia, Actinomyces
Semiinvasive Aspergillosis
= CHRONIC NECROTIZING ASPERGILLOSIS
= chronic cavitary slowly progressive disease in patients with preexisting lung injury (COPD, radiation therapy), mild immune suppression, or debilitation (alcohol, diabetes)
• symptoms mimicking pulmonary tuberculosis
√ progressive consolidation (usually upper lobe)
√ development of air crescent and fungus ball over a period of months
Dx: pathologic examination demonstrating local tissue invasion
Invasive Pulmonary Aspergillosis
= often fatal form in severely immunocompromised patients with absolute neutrophil count of < 500
Predisposed: most commonly in lymphoma / leukemia patients with prolonged granulocytopenia; after organ transplantation
Path: endobronchial fungal proliferation followed by trans- bronchial vascular invasion eventually causes widespread hemorrhage + thrombosis of pulmonary arterioles and ischemic tissue necrosis with systemic dissemination; fungus ball = devitalized sequestrum of lung infiltrated by fungi
• history of series of bacterial infections + unremitting fever
• pleuritic chest pain (mimicking emboli)
• dyspnea, nonproductive cough
• progression of pulmonary infiltrates not responding to broad-spectrum antibiotics
(a) early signs
Frequency: 96% (19%) on day 0 (day 14)
√ single / multiple ill-defined peripheral opacities abutting the pleural surface
√ “CT halo” sign = single / multiple 1–3-cm peripheral nodules (= necrotic lung) with halo of ground-glass attenuation (= hemorrhagic edema)
√ patchy localized bronchopneumonia
(b) later signs of progression
√ enlargement of nodules into diffuse bilateral consolidation
√ development into large wedge-shaped pleural-based lesions
√ “air-crescent” sign (in up to 50%) = cavitation of existing nodule (air crescent between retracting sequestered necrotic tissue and surrounding rim of hemorrhagic lung parenchyma) 1–3 weeks after recovery from neutropenia
◊ has better prognosis than consolidation without cavitation (= feature of resolution phase)
Prognosis: mortality rate of 50–90%
Dx: biopsy showing branching hyphae at tissue examination; sputum culture positive in only 10%
Rx: amphotericin B
Allergic Bronchopulmonary Aspergillosis
= ABPA = hypersensitivity reaction to aspergillus antigens released by colonization of tracheobronchial tree by Aspergillus fumigatus in patients with long-standing asthma / cystic fibrosis
◊ Most common + clinically important form of aspergillosis!
Prevalence: in 2–32% of patients with asthma; in 2–15% of patients with cystic fibrosis
Age: mostly young patients (begins in childhood); may be undiagnosed for 10–20 years
Path: bronchocentric granulomas within bronchi and bronchioles with associated mucoid impactions
Histo: deposition of immune complexes + inflammatory cells in bronchial mucosa → necrosis + eosinophilic infiltrates
Pathogenesis: IgE-mediated type I hypersensitivity reaction + specific IgG-mediated type III hypersensitivity
Pathophysiology:
inhaled spores trapped in segmental bronchi of individuals with asthma → germinate and form hyphae → aspergillus antigen reacts with IgG antibodies → immune complexes activate complement → release of proteolytic enzymes → inflammatory infiltrates → bronchial wall damage → central bronchiectasis
Staging (Patterson):
I acute phase with all primary diagnostic criteria
II remission = clearing of pulmonary infiltrates with declining IgE levels
III exacerbation = all criteria of stage I reappear after remission
IV corticosteroid dependency
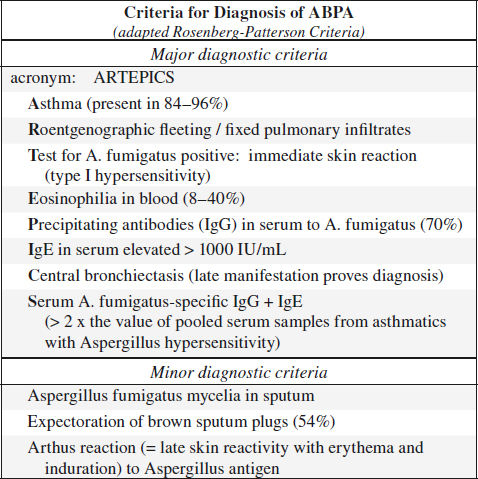
V irreversible lung fibrosis
A. ACUTE ABPA
Type I reaction = immediate hypersensitivity (IgE-mediated mast cell degranulation)
Histo: alveoli filled with eosinophils
• bronchoconstriction, mucus production
• bronchial wall edema ← ⇑ vascular permeability
B. CHRONIC ABPA
Type III reaction = delayed immune complex response = Arthus reaction (IgG-mediated)
Histo: bronchocentric granulomas + mucoid impaction; fungal hyphae without tissue invasion
• flulike symptoms: fever, headache, malaise, weight loss, fleeting pleuritic chest pain
• wheezing, expectoration of brown mucous plugs
• positive skin test to A. fumigatus; peripheral eosinophilia
• elevated IgE level correlates well with disease activity!
CXR:
√ initial CXR normal in up to 50%
√ NORMAL peripheral bronchi
√ hyperinflation ← bronchospasm / emphysema
(a) early stage:
√ migratory pneumonitis = ill-defined homogeneous transient recurrent “fleeting” alveolar patchy subsegmental / lobar opacities (in up to 90%)
Location: upper lobes (50%), lower lobes (20%), middle lobe (7%); both lungs (65%)
Duration: may persist for > 6 months
(b) later stage of bronchial wall damage:
√ central varicose / cystic bronchiectasis with thickened bronchial walls:
√ “tramline” / parallel line bronchial walls ← edema
√ 1–2-cm ring shadows (= bronchus on end)
√ “finger in glove / gloved finger / toothpaste shadow” = V- or Y-shaped branching and tapering tubular opacities arising centrally and extending peripherally remaining for months + growing in size
Location: perihilar + upper lobes
Path: plugging of airways by hyphal masses with mucoid impaction distally in 2nd order bronchi of 2.5–6 cm in length
√ perihilar opacities simulating hilar and/or mediastinal lymphadenopathy ←mucus-filled dilated central bronchi
√ isolated lobar / segmental atelectasis (in 14%) with collateral air drift
√ lobar consolidation (in 32%)
√ cavitation (in 14%) ± air-fluid levels ← postobstructive / eosinophilic abscess
√ UNUSUAL: aspergilloma in cavity (7%), empyema, pneumothorax
(c) chronic stage:
√ pulmonary fibrosis + retraction
√ hilar elevation ← lobar shrinkage
CT:
√ bronchiectasis of segmental + subsegmental bronchi affecting > 3 lobes
√ CHARACTERISTIC highly opaque mucus within dilated bronchi ← calcium salts / metals (iron and manganese) / desiccated mucus / calcifications (in 30%)
√ tree-in-bud appearance = centrilobular nodules connected to branching linear structures (more common in asthmatics)
√ patchy areas of mosaic attenuation ← concomitant small-airway disease + areas of air trapping
√ atelectasis, consolidation, air trapping
Prognosis: end-stage pulmonary fibrosis → respiratory and right heart failure (if untreated)
DDx: hypersensitivity pneumonitis or allergic asthma (no hyphae in sputum, normal levels of IgE + IgG to A. fumigatus), tuberculosis, lipoid pneumonia, Löffler syndrome, bronchogenic carcinoma
Cerebral Aspergillosis
Transmission:
(1) hematogenous dissemination from distant pulmonary infection
(2) angiotropic / perineural spread from paranasal sinus / orbital infection (rhinocerebral disease)
(3) direct traumatic implantation
√ ring-enhancing cerebral abscesses ← hematogenous dissemination
√ hypoattenuating intracavitary projections without associated enhancement on T2WI + ADC map ← proliferating hyphae
√ propensity for vascular invasion by fungal hyphae ← production of elastase
√ zones of low T2 signal intensity ← iron, manganese, magnesium in paranasal fungal concretions
Cx: cerebral infarction, hemorrhage (25%), mycotic aneurysm ← invasion of vessel wall
Prognosis: in immunocompromised ~ 100% mortality
Pleural Aspergillosis
= Aspergillus empyema in patients with pulmonary tuberculosis, bacterial empyema, bronchopleural fistula
√ pleural thickening
Renal Aspergillosis
= uncommon renal infection
Renal aspergillosis occurs in immunocompromised patients with diabetes / HIV infection / on corticosteroid therapy.
Route: hematogenous dissemination (most common) / ascending infection / Aspergillus casts in renal pelvis
√ mimicks complex renal mass / cyst / abscess
CT:
√ hypoattenuating mass with thick enhancing wall
√ internal septations
√ ± features of focal pyelonephritis in surrounding renal parenchyma with delayed enhancement
Dx: urinalysis, aspiration of lesion
ASPIRATION OF SOLID FOREIGN BODY
In suspected foreign-body ingestion the initial standard imaging protocol includes frontal and lateral radiographs of the chest, neck (often included on CXR) and abdomen.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


