Congenital heart disease (CHD) can broadly be thought of as what happens when normal heart development goes awry. Although the range of defects may seem endless, there are limitations. Pathologists, cardiologists, and cardiothoracic surgeons have successfully developed systems for the nomenclature, definition, and classification of CHD. Accordingly, the chapters of this book are organized by cardiac lesions. This systematic approach is possible only because there are two significant restraints on what leads to CHD. One is the progression of cardiac development. The heart forms through a sequential series of embryological events. The effects of certain events going wrong will not be observed unless prior events were successfully completed. For example, you cannot have abnormal looping of the heart tube without the tube itself first forming. The second constraint is viability. Defects that are incompatible with intrauterine life do not get the attention of the cardiac surgeon or cardiologist and may provide only cardiac pathologists with topics of academic discussions. As imaging technologies have improved, a greater range of disease has been seen by fetal cardiologists and sonographers. Now, fetal echocardiography may be performed at earlier stages of gestation and can reveal structural defects that would not prove viable later in development. In addition, new insights continue to be made about the progression of CHD during development. This adds increased onus on those diagnosing and treating patients prenatally to understand normal cardiac developmental biology and how the embryological processes may be perturbed. Many key events in cardiac development are complete before imaging can be performed, but advancing technology brings us ever closer to being able to observe these events in patients ( Figure 2-1 ).

Early Cardiogenesis
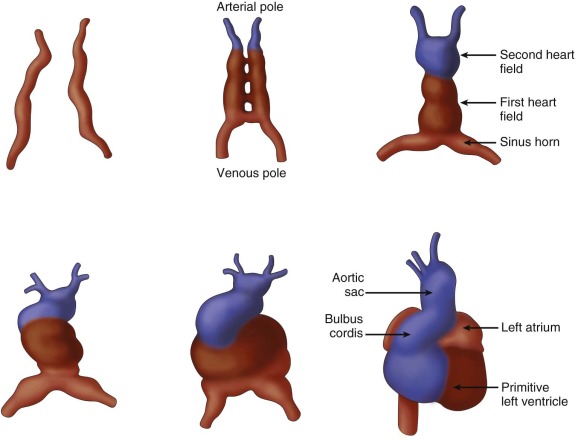
The earliest steps in the formation of the heart start at the time of gastrulation, which is the formation of the three germ layers—ectoderm, endoderm, and mesoderm. A subset of cells from the mesoderm layer will give rise to the bulk of the heart, and these cells make up the cardiogenic fields. They arise on the two sides of the midline and meet in the middle at the anterior part of the embryo to form the cardiac crescent ( Figure 2-2 ). Recent work has shown that the cardiogenic fields can be subdivided into two groups—the first heart field and the second heart field (sometimes referred to as the posterior and anterior heart fields), which, in turn, will form the left and right myocardium, respectively. Thus, before the ventricles themselves form, there is already a molecular basis for differences between the right and the left ventricular myocardium. The two sides of the cardiac crescent fuse along the midline to form the primitive heart tube. The primitive heart tube can itself be subdivided into regions along the caudal to rostral axis: sinus venosus, primitive atria, primitive ventricle, bulbus cordis (conus), and truncus arteriosus. The primitive heart tube begins to contract in a peristaltic manner at approximately 5 weeks’ gestation.

Cardiac Looping
As the primitive heart tube develops, it folds on itself and twists in a process called looping ( Figure 2-3 ). The mechanism that underlies this process continues to be debated, but one recent hypothesis that has gained favor is that looping results from differential ballooning out of the chambers, rather than rotational movement of the cells. Normally, the looping occurs to the right and results in a d -looped heart. In some cases of CHD, looping may occur to the left ( l -looped). The process of looping is the first visible sign of left-right asymmetry apparent in the developing embryo, although genes involved in this process have been shown to be differentially expressed before this process occurs. Looping sets up the relationship between the inflow tract, the outflow tract, and the ventricular septum of the right ventricle, which is important in the nomenclature of CHD.
Septation
In mammals and birds, the pulmonary and systemic circulations are separate; they are in series with one another in adults. In order for this to occur, the atria, atrioventricular (AV) valves, the ventricles, and the outflow tract must be divided during development.
Atrial and Canal Septation
Because atrial and canal septation are linked, they are discussed together. The atria are the first structures to begin to septate, and the last to finish, with the foramen ovale not closing under normal conditions until after birth (and then remaining probe patent for some time). At the beginning of the sixth week of gestation, the pulmonary venous confluence (see later) evaginates into the roof of the embryonic atrium between the two growing atrial appendages ( Figure 2-4 ). Cranial to this, the septum primum (primary atrial septum) forms as a muscular septum in the shape of a crescent. It grows from the dorsal wall of the atrium toward the AV canal. It has been described as completely dividing the atria first (hence, primum) and then later becoming perforated to form the foramen ovale. However, it likely never fully closes, because blood needs to flow from the right atrium to the left throughout development. The septum secundum arises along the rim of the pulmonary vein as a structure called the dorsal mesenchymal protusion (also called the atrial spine, spina vestibule, or vestibular spine). It has been appreciated that this consists of both atrial cells as well as “extracardiac mesenchyme” that migrates in from the dorsal attachment of the heart to the body. The septum secundum contributes to the division of the AV valve to allow formation of separate tricuspid and mitral valves. Defects in the formation of the dorsal mesenchymal protrusion lead to the formation of a common AV canal in the most extreme cases and to an atrial septal defect in more mild cases. Confusingly, defects in the septum secundum result in “primum” atrial septal defects. Conversely, defects of the septum primum are called “secundum” atrial septal defects. The thin, membranous septum primum forms to the left of the more muscular septum secundum and functions as a flap valve allowing right-to-left flow. Postnatally, when the pressure in the left atrium becomes higher than that of the right, the flap closes the foramen ovale to complete the septation of the atria.

Ventricular Septation
Following normal looping, the primitive right and left ventricles are positioned relatively rightward and leftward to each other ( Figure 2-5 ). It is important to remember that they are not at the same level in the anteroposterior plane. The primitive right ventricle is more anterior. The flow of blood comes into the left ventricle, then goes across the bulboventricular foramen to the right ventricle and out the as-yet-undivided outflow tract. As development progresses, inflow becomes more directed toward both ventricles. (Failure of this process can result in a double-inflow left ventricle [DILV]—a situation much more common than double-inflow right ventricle). The ventricular septum begins to grow toward the AV canal and outflow tract from the apical and inferior portion of the junction between the primitive right and left ventricle. This forms the muscular part of the interventricular septum. Incomplete growth during this stage can result in muscular septal defects. Septation of the ventricle is complete when the muscular septum meets the canal septum between the AV valves and the conal septum just below the now separate outflow tracts. If the canal septum has not formed properly, a canal type ventricular septal defect may be left. Similarly, if the conal septum forms to far anterior or posterior, the muscular septum may not fuse with the conal septum, causing a septal malalignment defect. Finally, if the conal septum forms normally but there is incomplete fusion between it and the muscular septum, a conoventricular defect results. In the area at which these structures meet, there is the thinner membranous septum.

Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


