Table 16.1 Constitutional Genetic Abnormalities Associated with Adrenal Cortical Tumours (ACT) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
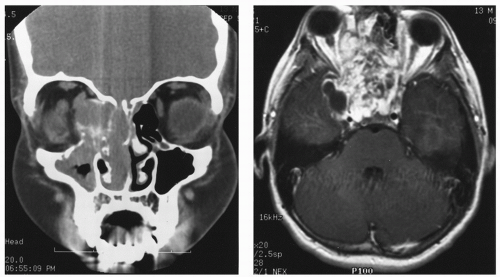 Figure 16.1 Coronal computed tomography (left) and axial magnetic resonance imaging (MRI) (right) of a 13-year-old boy with headaches, diplopia, and right proptosis. The mass-produced destruction of the ethmoid air cells and superior turbinates, mass effect in the right orbit, compression of the right cavernous sinus, displacement of the right internal carotid artery, and displacement of the pituitary. A biopsy proved the diagnosis of juvenile nasopharyngeal angiofibroma. Three-dimensional treatment planning was used to prepare a four-field noncoplanar external beam radiotherapy treatment technique using rigid head immobilization. A dose of 36 Gy at 2 Gy per fraction was administered. One and one-half years after irradiation the symptoms had significantly improved and the mass was smaller on physical examination and MRI. Shortly thereafter the symptoms recurred, and the MRI demonstrated regrowth of the angiofibroma, within and outside the irradiated field. Interferon was administered as antiangiogenesis agent. |
primary malignancies is observed in other studies, and it points out the need for close surveillance especially after RT (19).
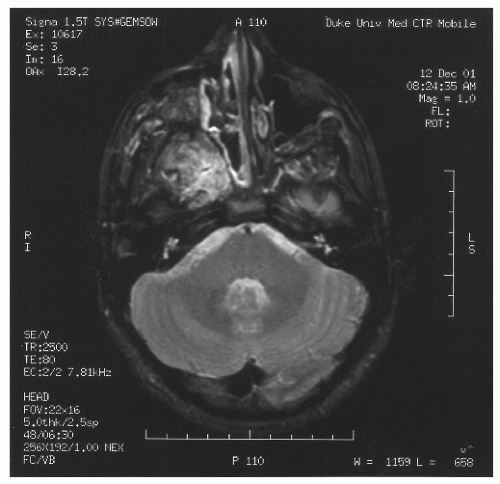 Figure 16.2 A 14-year-old boy presented to the radiation oncology department after multiple surgical and embolization procedures intended to remove and devascularize a juvenile nasopharyngeal angiofibroma. Detailed three-dimensional mapping of the tumor was performed using magnetic resonance imaging accompanied by careful fiber-optic examination. The mass was treated, with a margin, with a mixture of 6- and 15-MV photons to a cumulative dose of 40 Gy. Over the subsequent 2 years, striking regression of the mass was seen. Before RT, the child had been subjected to repeated episodes of bleeding, which ceased after RT. |
Carcinoma derived from the follicular epithelium:
Follicular carcinoma: no papillary nucleus are seen in these tumors; capsular and vascular invasion is common
Papillary carcinoma: nucleus are typical, with cytoplasmic inclusions
Poorly differentiated carcinoma: includes insular and solid/trabecular carcinoma, with morphology between well-differentiated and poorly differentiated tumors
Nondifferentiated carcinoma: loss of follicular architecture, with aggressive behavior (necrosis, mitosis, vascular invasion, ….)
Medullary carcinoma: this subtype derived from C-cells or neural crest—parafollicular cells.
Other tumors: lymphomas and metastasis from other cancers
differentiated thyroid cancers were multifocal at diagnosis. Lung is the more frequent site of distant metastasis.
confirms that outcome of pediatric thyroid carcinoma is independent of prognostic factors often used in adults: extrathyroid invasion, metastasis, and tumor size (43).
Table 16.2 Differentiated Thyroid Cancer in Children from 11 Pediatric Series | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
spaces is limited only by bone, which can be eroded by pressure. The vascular structures of the basisphenoid region can be involved, especially in the area of the sphenopalatine foramen (80). Intracranial extension has been reported in 10%-20% of all cases before the era of modern neuroimaging. JNA’s etiology and pathogenesis remain unknown. Origin of JNA are discussed: main retained causes are vascular or fibrous.
complications (1 patient vs. ≥30 patients), shorter length of hospital stay (2 vs. 5 days), and lower rate of recurrence (0% vs. 24%) (116). In long-term results of the endoscopic approach, a retrospective study of 21 consecutive patients undergoing endoscopic resection of JNA showed, with a mean follow-up of 52 months (range of 5-120), 71.4% of the patients were free of disease after one endoscopic resection (117). Three patients (14.3%) developed a recurrence with the need for further treatment at 6, 14, and 23 months, respectively. Two of the three recurrent tumors were successfully resected endoscopically; one case was treated with gamma knife.
Table 16.3 Six Proposed Staging Systems for Juvenile Nasopharyngeal Angiofibroma | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
literature. Recently, McAffee et al. took approximately 2-cm margins around the tumor for the treatment planning with excellent local control (76). Late effects, especially the risk of malignant cancer, neuroendocrine dysfunction, cataract …, must always be considered but these effects remain quite rare and should be weighted against the risk of surgery in case of advanced disease. A large retrospective study on 130 patients treated over a 41-year period (1960-2000) was published. Twenty-seven patients received radiation (30-55 Gy) as first treatment (121). Fifteen percent of the irradiated patients developed relapse 2-5 years later. Fifteen percent of the patients developed significant late complication, including growth retardation, panhypopituitarism, temporal lobe necrosis, cataracts, and radiation keratopathy. Two of the patients developed in-field cutaneous basal cell carcinomas but were at risk for skin tumors. Second malignancies have been observed in other series. Cummings et al. reported one patient who developed thyroid carcinoma after 35 Gy (124). The main visual complication after RT is cataract formation, while optic neuropathy and retinopathy are uncommon at conventionally fractionated doses between 30 and 36 Gy. Cummings et al. reported cataract development in 2 of 55 patients (3.6%) (124). Lee et al. observed cataracts in 3 patients on 27 (11%) (121). Cummings examined the comparable risk factors associated with surgery and RT and found that the relative risks of significant complications were similar with both treatment modalities (124). Intracavitary RT has been used rarely (125) but not used anymore due to major bleeding and inadequate coverage of the treatment volume (124).
Table 16.4 Radiotherapy for Juvenile Nasopharyngeal Angiofibroma: Most Recent Studies | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


