Endocrine, Metabolic, and Nutritional Diseases
Endocrine Disorders
Acromegaly
Background
Acromegaly is marked by the oversecretion of growth hormone (somatotropin) in a skeletally mature patient. Oversecretion is caused by an adenoma in the anterior lobe of the pituitary. Anterior pituitary tumors account for about 18% of all intracranial tumors. The incidence of acromegaly is reportedly 50 to 60 cases per million and the prevalence is 3 to 4 cases per million per year.2 Acromegaly has no gender bias. If the level of growth hormone increases, it produces overgrowth of bone, particularly those bones that are formed intramembranously, principally the skull and mandible. Enlarged joint spaces and soft-tissue swelling are typical, producing enlarged hands and feet with thickening of the tongue.
Because the growth centers have not closed, increased levels of growth hormone in skeletally immature patients result in giantism.2,68,71 Giantism patients with persistent increases in growth hormone also demonstrate acromegalic features. Complications include degenerative joint disease (DJD), increased risk for colonic polyps and colon cancers, and death caused by cardiovascular and cerebral vascular disease. Patients who exhibit diabetes mellitus or hypertension have the highest mortality rate.2,68
Imaging Findings
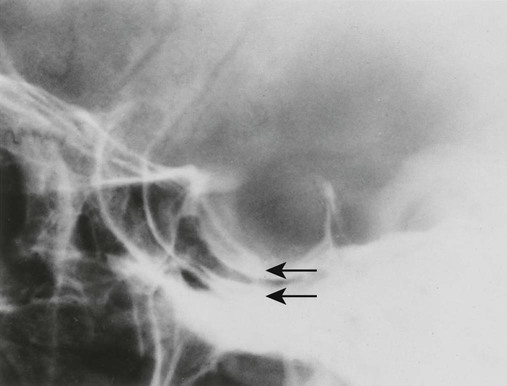
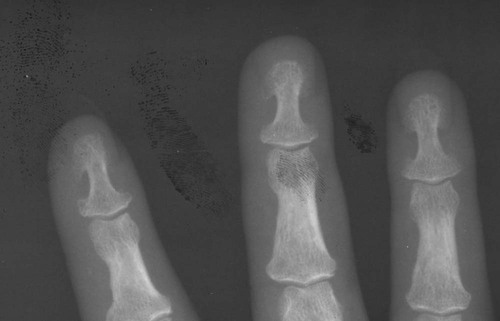
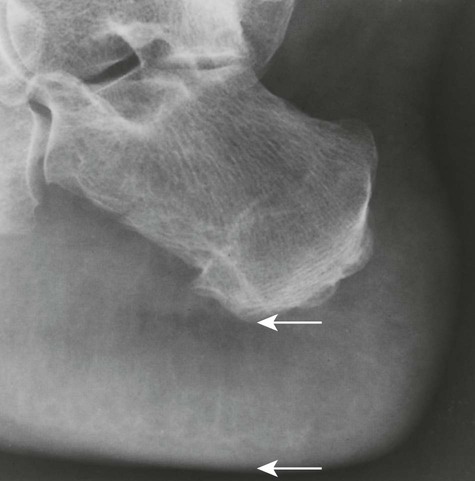
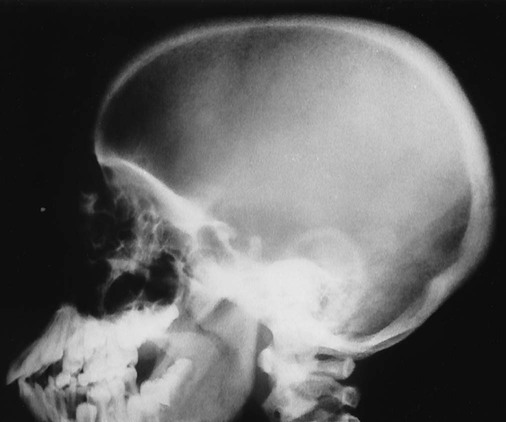
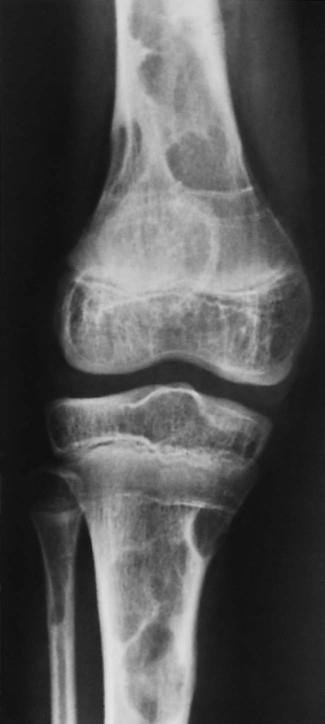
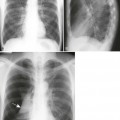
Increased disc spaces and other joint spaces are demonstrated early. This is followed by generalized osteoporosis and DJD in advanced cases. A longitudinal study followed subjects for an average of 14 years and observed osteophytosis in the presence of modest joint space narrowing. This study suggests growth hormone (GH) mainly stimulates bone formation while possibly protecting against cartilage loss.77 Increased anteroposterior (AP) diameter of the thorax and enlargement of the ribs at the costochondral junction is typical.2,68,71 Sella turcica expansion (Fig. 14-1) or destruction, enlarged sinus cavities, malocclusion, and widened mandibular angle (prognathism) are common. The hands and feet exhibit thickening of the tubular bones and prominent ungual tufts (Fig. 14-2). Soft-tissue thickening may be seen at the heel pad (Fig. 14-3) measuring 20 mm or more.2,68 Although this measure varies, the heel pad thickness should not exceed 23 mm among female and 25 mm among male patients.
Clinical Comments
Acromegaly is a gradually progressive disorder in which the symptoms often precede diagnosis by 5 to 10 years. The classic features are a prominent forehead, malocclusion associated with a wide mandibular angle, thickening of the tongue, and large hands and feet. Soft-tissue swelling may produce neural compression in the carpal tunnel. Barrel chest, seborrhea, sweating, hypertrichosis, hyperglycemia, hypertension, and cardiomyopathy are frequently occurring features.2,68,71 Diagnosis is accomplished by documenting excess GH secretion. Some are suggesting a novel use of facial recognition software may be helpful in early detection and/or monitor the progression of disease.61 In cases of episodic excess of GH, documentation of oral glucose administration that fails to suppress GH secretion is helpful. Magnetic resonance imaging (MRI) or computed tomography (CT) documentation of pituitary hyperplasia is suggested.2,68 Treatment revolves around transsphenoidal surgical removal or destruction of the pituitary tumor by some form of radiation therapy, reversal of hypersecretion, and maintenance of normal anterior and posterior pituitary function.2,57,68
Cushing Syndrome
Background
Cushing syndrome (CS) is hyperadrenocorticism secondary to anterior lobe pituitary tumors, tumors that secrete ectopic adrenocorticotropic hormone (ACTH), adrenal cortex tumors, or as a complication of glucocorticoid therapy (Fig. 14-4). Cushing disease adds an additional component, hypothalamic–pituitary dysfunction. The incidence of adrenal tumors in the United States is 0.5 per million people per year.39 Pituitary tumors producing CS are more frequent.75 Women are more commonly affected, and a wide age distribution is seen.22,34,39 Growth retardation and suppressed sexual maturation are most notable in children. Additional characteristics include rapid weight gain associated with abnormal fat distribution. A thick layer of facial fat rounds out the cheeks, producing the typical circular “moon face” appearance. In addition, a prominent fat pad is deposited over the upper thoracic spine, producing a mass called a buffalo hump. The abdomen typically is pendulous. Patients also may demonstrate hypertension, rapid hair growth,19,49 muscular atrophy,33 and striae.39,49
Imaging Findings
The hallmark radiographic sign is generalized osteoporosis typified by marked loss of trabecular bone and less evident loss of cortical bone. Also evident is an increased number of insufficiency fractures of the vertebrae, scapula, ribs, and pubic bones.19,22,34,49,68 Corticosteroid therapy producing CS also may lead to osteonecrosis49,68 and insufficiency fractures.49
Clinical Comments
Excessive production of cortisol is caused by adrenocorticotropic hormone (ACTH) hypersecretion by pituitary adenomas, and occasionally by ectopic ACTH-secreting tumors or autonomous adrenocorticoid tumors.
Characteristic Features.
These features include facial-trunk obesity (buffalo hump, moon face), abdominal striae, growth retardation, rapid hair loss, and muscular atrophy. Generalized osteopenia, insufficiency fractures, and occasional osteonecrosis are the primary imaging findings.19,22,23,34,39,75 Enlarged sella turcica or evidence of pituitary hypertrophy29 may be present but is not a routine finding.9 Plain films of the skull, chest, and abdomen often are normal. CT, MRI, and ultrasound often are necessary.9,40 Angiography and venous sampling are useful to document the hypovascularity that is typical of endocrine tumors.40,75 Occult secretory tumors may be found with scintigraphy.33 In cases of recurrent Cushing disease, the treatment of choice is transsphenoidal pituitary adenectomy. Surgery is followed by irradiation (4000 to 5000 rad). Surgical success rate is 90% for skilled neurosurgeons.38 Hypercortisolemia, external pituitary irradiation, and posttreatment hypopituitarism may increase the risk of brain infarction.42
Giantism (Gigantism)
Background
Hypersecretion of GH occurring in a skeletally immature patient produces excessive proportional bone growth, known as giantism. This unusual condition is produced typically by adenomas of the anterior lobe of the pituitary gland or, less commonly, by diffuse hyperplasia of acidophilic cells elsewhere. Organomegaly and commensurate hypertrophy of muscles and connective tissue are associated findings.
Imaging Findings
The hallmark imaging findings of true giantism are of proportional, yet exaggerated, skeletal growth. The bones are increased in both length and diameter.5,17 Enlargement of the sella turcica or evidence of pressure erosion may be seen occasionally on plain film or, more reliably, with CT. MRI better demonstrates the abnormal pituitary gland. Combinations of imaging may be necessary to identify ectopic sources of GH.70
Clinical Comments
Remarkable proportional growth is typical. The skeletal maturation of patients with giantism may exceed that of their age-related peers by 11 standard deviations. A case study reported a 3.5-year-old boy who demonstrated a level of skeletal maturity consistent with a 10.5-year-old child. At 7 years of age he was a well-proportioned boy 182 cm (6 feet, ¾ inches) tall, weighing 99.4 kg (219 lb) and wearing size 13EEE shoes.70 Laboratory profiles include elevations of 24-hour GH secretion, paradoxic growth response to administered thyrotropin-releasing hormone (TRH), and failure to suppress serum GH levels with oral glucose loading.5,17,70 Hypercalcemia, hyperphosphaturia, and elevated levels of alkaline phosphatase are consistent findings.
Hyperparathyroidism
Background
Hyperparathyroidism (HPT) is the condition of elevated levels of parathormone (PT), resulting from a variety of direct and indirect stimuli.16,68,69 Excess levels of PT produce disorders of calcium, phosphate, and bone metabolism; this is the most common cause of hypercalcemia.67,69 HPT occurs in approximately 1 in 700 persons. HPT is divided into three categories: primary, secondary, and tertiary.
Primary HPT is most commonly produced by a single adenoma (80% to 85% of cases), less commonly, parathyroid hyperplasia (10% to 15% of cases), multiple adenomas (4% to 5% of cases), and, rarely, secretory carcinomas (1% to 3% of cases).23,69 Secondary HPT results from chronic renal failure in most cases (Fig. 14-5), representing an end-organ dysfunction.27,32,68 PT secretion is stimulated by elevated serum levels of phosphate and reduced ionized serum calcium.27,68 Diffuse enlargement of all parathyroid glands is typical.23 Tertiary HPT may be described as a complication of dialysis. Parathyroid glands may act independently of the serum calcium levels.27,68 In this setting, as serum phosphate levels increase, stimulus for PT secretion also increases.
PT acts predominantly on the skeleton, the kidneys and gastrointestinal tract to mobilize skeletal calcium into circulation, reducing the renal excretion of calcium and increasing gastrointestinal absorption of calcium by a diversity of metabolic actions.23,69 Although primary HPT remains the most common form of HPT, secondary and tertiary HPT are increasing in frequency. This is most likely a result of an increasingly larger geriatric population, more frequent encounters with the causes of secondary and tertiary HPT, and increased longevity. Primary HPT is a disease that affects predominantly middle-age women, half of whom are postmenopausal. The balance of the cases are equally divided between men and premenopausal women.16,23,67 It is rare to find patients younger than 16 years of age.69
Imaging Findings
It is now recognized that HPT is more common than previously thought,16,23,56,67,69 and is most likely to demonstrate little or no specific signs or symptoms.16,56,67 The insensitivity of plain film and earlier recognition of HPT by laboratory examination often produce essentially normal radiographs.56 Bone mineral density assessments using single- or dual-energy x-ray absorptiometry or quantitative CT are more useful and should be used to investigate asymptomatic HPT.16,56,68 Bone biopsy is definitive, but it is not used on a routine basis and is primarily a research tool.27,67
The radiographic analysis must answer two basic questions. First, what is the cause of the elevations of PT? Determining this establishes presurgical localization, treatment of renal disease if possible, and change in dialysis technique if necessary. Second, what is the clinical status and what skeletal and extraskeletal manifestations are present?
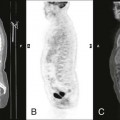
Follow-up examinations monitor successful treatments or identify progressive disease. Experts do not agree on which imaging strategy is best. Successful imaging often includes several complementary modalities. Because the majority of cases of primary HPT are caused by a single adenoma (80% to 85%), high-resolution ultrasound by an experienced sonographer is advocated as a first step. This study is considered safe, cost-effective, and relatively sensitive. Its value is diminished by small or ectopic lesions.23,60,67 Scintigraphy in the form of technetium-99m (99mTc)-sestamibi localizes parathyroid tumors normally situated and in ectopic locations. It also can determine parathyroid gland functionality.23,60,67 MRI is indicated in cases of HPT with essentially normal parathyroid glands or persistent postsurgical elevations of PT. This clinical picture often is associated with ectopic secretory tissue.23,60 Contrast-enhanced CT, arthrography, and selected venous sampling may be used in difficult cases or if reoperation is contemplated.40,60,69
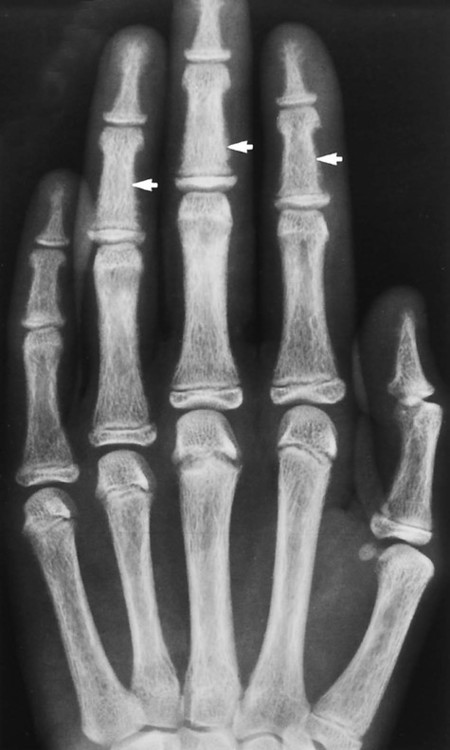
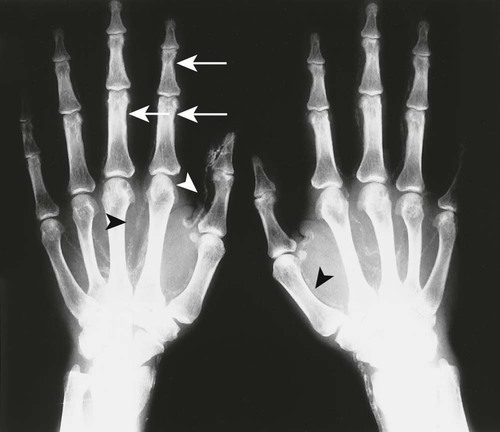
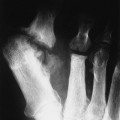
The effects of HPT on the skull and extraskeletal structures are numerous. Some are classically, if not exclusively, associated with HPT. HPT effects on bone probably include a higher remodeling rate of the haversian canals. Ultimately, longitudinal defects appear in cortical bone.27,37,56,69 Hyperthyroidism, Sudeck atrophy, and Paget disease may appear similarly. Cortical endosteal and trabecular resorption also occur.27 Subperiosteal reabsorption is seen early and most often found along the radial side of the middle phalanx of the hand, particularly the second and third digits. Fine-grain screens producing images with great detail18 are necessary for early recognition of this virtually pathognomonic sign.16,27,32,50,56,68,69 If progression of the disease occurs, ungual tufts (acroosteolysis), proximal phalanges, and metacarpals are involved (Fig. 14-6).27
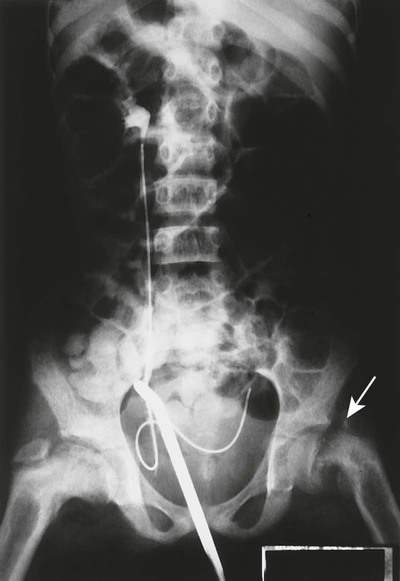
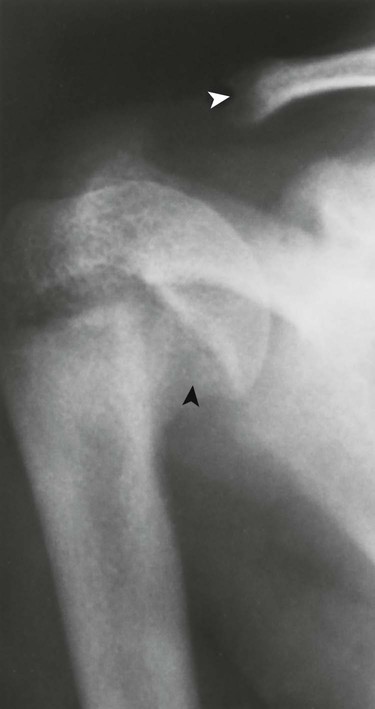
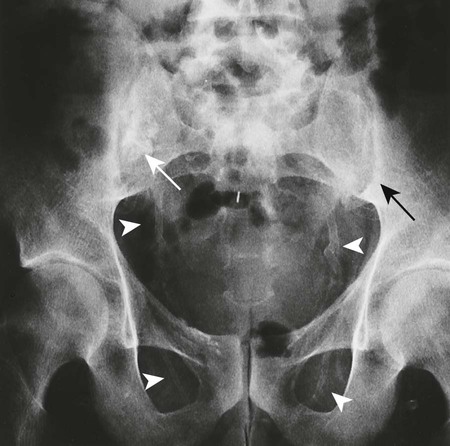
Reports exist of generalized osteopenia16 with accentuated trabeculae pattern caused by resorption of nonessential trabeculae and loss of cortical definition. The latter involves both periosteal and endosteal cortices. This produces blurring and irregular, thinned cortices with widened yet rarified medullary cavities.27,68 Pseudowidening of the joint space may be seen as resorption of the sacroiliac (Fig. 14-7), acromioclavicular (Fig. 14-8), and pubic joint surfaces.27,68 The upper medial surface of the humerus, tibia, calcaneus, ischial tuberosities, and the upper border of the ribs may demonstrate resorption.27 The fine, permeative, destructive pattern of the skull, known as “salt-and-pepper” skull, is also typical of HPT (Fig. 14-9).66,68 Resorption of the lamina dura surrounding the tooth socket also is seen. Administration of vitamin D and calcium leads to accelerated loss of bone mineral density in some patients, and cases of hyperphosphatemia exhibit increased incidence of soft-tissue calcification.
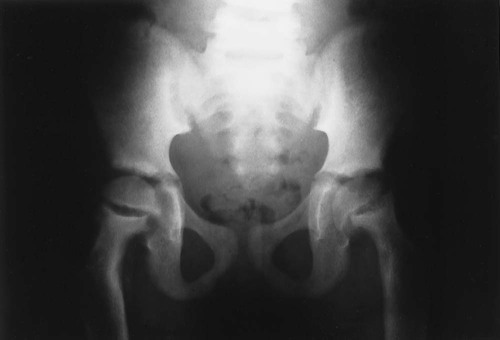
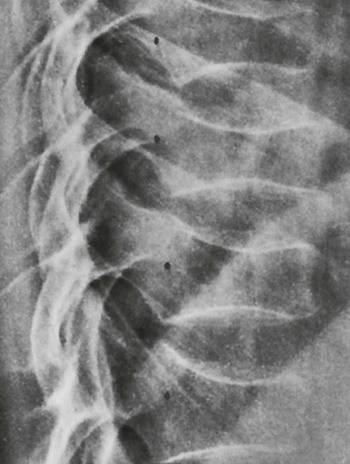
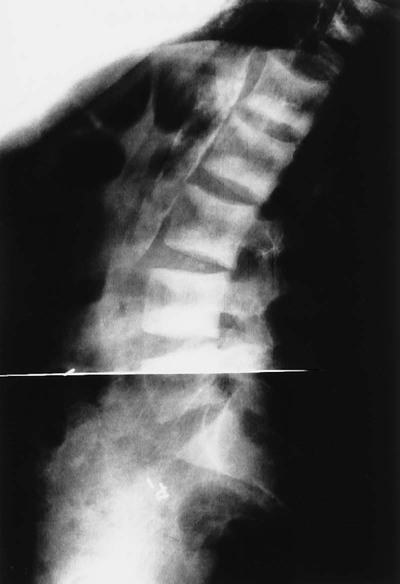
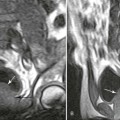
Osteitis fibrosa cystica, also called a brown tumor (Fig. 14-10), is a collection of fibrous tissue and giant cells in bone that is found in primary and secondary HPT.9,23,69 These present as geographic lucencies. Slightly expansile, they are most commonly found in the mandible, pelvis, ribs, and femora.22 Overall, brown tumors are seen infrequently.67 Soft-tissue calcifications are seen in later stages of HPT (Figs. 14-11 and 14-12).27,68 Nephrocalcinosis is still considered to be a common presenting complaint.67 Chondrocalcinosis of menisci in the knee, as well as other sites, is seen. Secondary HPT is thought to produce more frequent soft-tissue calcifications of vascular and periarticular structures.27,40,68 Osteosclerosis of the spine (“rugger jersey” spine) (Fig. 14-13) is a characteristic, yet unusual, sign.14,68 Periarticular condensation of bone may be seen in the presence of open growth plates.14
Clinical Comments
As our understanding of HPT has increased, it has become recognized that the most common presenting sign is hypercalcemia.16,27,32,50,56,68,69 Hypercalcemia in the presence of elevated PT by immunoradiometric assay (IRMA) or immunochemoluminescent assay (ICMA) is a definitive diagnosis.67 Some patients have persistent high normal levels of serum calcium. If this resulted from anything other than primary or secondary parathyroid disease, the PT levels would be low or nonexistent.67 Primary HPT may copresent with vitamin D deficiency; when both features are present this represents an “aggravated form of the disease.”43 Otherwise a paucity of presenting features exists.9 Silverberg and Bilezikian found that half of their patients were asymptomatic or demonstrated nonspecific complaints.67 These may include depression, subjective weakness, memory and sleep abnormalities, constipation, “bone pain,” loss of appetite, nausea, vomiting, or polydipsia.60,67,69 Some report an association between childhood irradiation of the neck and primary HPT.67 The classic features of HPT are decreased bone density, brown tumors, leontiasis ossea, renal calculi, chondrocalcinosis, and metastatic calcification. These features, although classic, are uncommon presenting features. Of these, renal calculi are the most frequent finding, occurring in 20% of all cases.32,67
An international panel suggests measurement of serum concentrations of 25-hydroxyvitamin D in suspected cases of primary HPT, and if below 20 ng/mL, to begin vitamin D supplementation prior to medical or surgical intervention.43
Hypoparathyroidism
Background
Hypocalcemia secondary to low or absent PT is the hallmark of hypoparathyroidism. Hypoparathyroidism most commonly follows thyroidectomy or radical laryngeal surgery.17 Cases of hypoparathyroidism have followed radioactive iodine treatments for HPT.10 Reports exist of metastases (typically breast cancer) and transient forms relating to surgical cure for HPT.14 Sporadic idiopathic and familial idiopathic conditions have a more variable presentation. Familial forms may be associated with pernicious anemia and hypoadrenalism. This is characterized further by circulating antibodies to the parathyroid, thyroid, and adrenal glands, supporting the concept of an autoimmune etiology in some cases.54,76 The earliest age of onset is seen in the familial form, often within the first year of life. Sporadic forms are typically found in patients 5 through 10 years old. The acquired form of hypoparathyroidism occurs later in life.
Imaging Findings
Generalized or localized sclerosis is the most common skeletal abnormality. Thickened calvaria and hypoplastic dentition, radiodense metaphyseal bands seen in the long bones, iliac crest, and vertebral margins also are potential findings. Occasionally reported are osteopenia, typically in pseudohypoparathyroidism. Asymptomatic periarticular calcifications, particularly those involving the hips and shoulders, are not rare.54
Clinical Comments
The predominant features of hypoparathyroidism evolve as a result of chronic hypoglycemia. These include tetany, epilepsy, cataracts, and papilledema with elevated intracranial pressure. Common yet not absolute features include short stature and mental impairment. Alopecia occurs in varying degrees. Underdevelopment of the dental roots is a common presenting complaint. Excessive renal reabsorption of phosphorus as a consequence of hypocalcemia is inevitable. Hypocalcemia and hyperphosphatemia combine to suppress 1,25-dihydroxyvitamin D synthesis to low levels.
Effective treatment includes calcium and vitamin D analogs. Serum calcium levels rise once the hyperphosphatemia reduces. Candidiasis of the nail beds or orogenital regions occurs in some sporadic or familial cases.5,17
Pseudohypoparathyroidism
Background
Pseudohypoparathyroidism (PHP) is an X-linked genetic disorder associated with normal or enlarged parathyroid glands and parathyroid target organ (bone, kidney) insensitivity. Often but not always, a typical skeletal phenotype occurs in addition to PT resistance and is called Albright hereditary osteodystrophy (AHO) type I.5,17
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


